Productos de oxidación del etileno
(Ir a página principal http://organica1.org/ )
Productos de oxidación del etileno
4.1.1. Óxido de etileno por
el proceso de la clorhidrina
4.1.2. Óxido de etileno por
oxidación directa
4.1.2.2. Realización del
proceso
4.1.2.3. Posibilidades de
desarrollo de la obtención de óxido de etileno
4.2. Productos derivados del
óxido de etileno
4.2.1. Etilenglicol y
etilenglicoles superiores
4.2.1.1. Posibilidades de desarrollo de la obtención de
etilenglicol
4.2.1.2. Aplicaciones del etilenglicol
4.2.1.3. Productos derivados: glioxal, dioxolano, 1,4-dioxano
4.2.3. Etanolaminas y derivados
4.2.5. Carbonato de 1,2-glicol
4.3.1. Acetaldehído por oxidación de etileno
4.3.1.2. Realización del proceso
4.3.1.3. Tendencias de mejoramiento
4.3.2. Acetaldehído a partir de etanol
4.3.3. Acetaldehído por oxidación de alcanos C3/C4
4.4 Productos derivados del acetaldehído
4.4.1.1. Ácido
acético por oxidación de acetaldehído
4.4.1.2. Ácido acético por oxidación de alcanos y alquenos
4.4.1.3. Acético por carbonilación de metanol
4.4.1.4. Posibilidades de desarrollo en la obtención del
ácido acético.
4.4.1.5. Aplicaciones del ácido acético
4.4.2. Anhídrido acético y cetena
4.4.3. Condensación aldólica del acetaldehído y productos
derivados
El óxido de etileno (oxirano) y el acetaldehído
son los productos de oxidación parcial e isómeros más sencillos del etileno.
Corresponden, por su facilidad de reacción y por sus numerosos productos
derivados de los mismos de importancia industrial, a los productos intermedios
principales que se obtienen del etileno. No obstante, ambos consumen bastante
menos etileno que el que se destina a polietileno, e incluso en Europa
occidental al que se dedica a la producción de cloruro de vinilo como se
muestra en la tabla siguiente de usos del etileno en Alemania Federal, Europa
occidental y EE.UU.:
|
Producto |
Alemania1976 |
Japón 1977 |
Europa 1975 |
1977 |
19821) |
1976 |
EE.UU. 1977 |
19801) |
|
Polietileno
(alta presión y baja presión) |
52 |
42 |
52 |
55 |
54 |
42 |
43 |
46 |
|
Cloruro de
vinilo |
12 |
17 |
18 |
18 |
19 |
12 |
12 |
12 |
|
Oxido de
etileno y derivados |
12 |
13 |
13 |
11 |
11 |
21 |
18 |
19 |
|
Acetaldehído
y derivados |
11 |
n.p. |
7 |
n.p. |
n.p. |
3 |
3 |
3 |
|
Etilbenceno/Estireno |
6 |
8 |
7 |
7 |
7 |
8 |
9 |
8 |
|
Otros (p.
Ej., etanol, acetato de vinilo, alcoholes y olefinas lineales,
1,2-dibromoetano), cloruro de etilo, etilenimina, aldehído propiónico) |
7 |
20 |
3 |
9 |
9 |
14 |
15 |
15 |
1) Estimado.
n.p. = no publicado.
Tabla 4-1.
Usos del etileno en Alemania, Europa, Japón y EE.UU. (en %).
4.1 Óxido de
etileno
El óxido de
etileno, desde su descubrimiento por A. Wurtz en 1859 y su primera obtención
industrial en 1925 por la UCC hasta hoy, ha tenido una expansión enorme en su
producción. La tabla del margen resume, por cuanto se sabe, las cifras de
producción de los países industriales más importantes. La capacidad mundial
alcanzó en 1977 aproximadamente los 6 millones de toneladas anuales.
Paralelamente
al desarrollo de la capacidad de producción
se modificaron las formas de
síntesis que partían del proceso histórico en dos etapas (con la clorhidrina
del etileno como producto intermedio), hasta llegar a lograr la oxidación
directa de etileno, mucho más económica en enormes instalaciones, con
capacidades hasta 360 000 toneladas al año. Así, por ejemplo, a mediados de los
años cincuenta, casi la mitad de la
producción de óxido de etileno en los EE.UU. provenía todavía de la clorhidrina
de etileno; a partir de 1975, toda la producción era exclusivamente por
oxidación directa del etileno.
4.1.1. Óxido de
etileno por el proceso de la clorhidrina
Aunque apenas
se utiliza hoy con el etileno, el proceso de la clorhidrina, en dos etapas,
para la epoxidación de olefinas inferiores se sigue utilizando para el propeno.
Transcurría a través de la clorhidrina de etileno, como producto intermedio que
no se aislaba y que por calentamiento con cal apagada se transformaba en óxido
de etileno:
![]()

La
selectividad en óxido de etileno alcanzaba aproximadamente el 80% (C2H4)
y era, por tanto, satisfactoria. De todas formas se perdía prácticamente todo
el cloro. Por cada 100 kg. de óxido de etileno se formaban 10-15 kg. de
1,2-dicloroetano, 7-9 kg. de 2,2’-diclorodietiléter y unos 300-350 kg. de CaCI2.
Las causas
principales de cambiar este proceso por una oxidación directa, fueron la gran
cantidad de productos químicos necesarios y, especialmente, el alto precio del
cloro que tanto influye en el costo total, así como también la considerable
contaminación de las aguas residuales.
4.1.2. Óxido de
etileno por oxidación directa
4.1.2.1 Fundamentos químicos
En 1931
consiguió T. E. Lefort la primera oxidación directa del etileno a óxido de
etileno, que luego, en 1937, la UCC pudo transformar en proceso industrial.
Posteriormente, participaron también otras empresas con perfeccionamientos para
su desarrollo, que principalmente fueron progresos en la obtención del
catalizador. No obstante, continúa siendo la plata el componente más activo y
selectivo de los catalizadores.
La oxidación
parcial del etileno con catalizadores de plata es una reacción exotérmica:

Pero está
acompañada principalmente por otras dos reacciones secundarias aún más exotérmicas, que son la combustión total del
etileno, que produce la cantidad principal de CO2 y la reoxidación
del óxido de etileno:
![]()

Los procesos
industriales alcanzan una selectividad del 65-70% en óxido de etileno, en los
que se produce un desprendimiento de calor de 110-120 kcal (503 kJ)/mol
etileno. Teniendo en cuenta el
mecanismo de reacción [ver ecuaciones
(6-8)], la selectividad máxima posible en óxido de etileno llega
sólo al 80%.
La activación
específica del O2 en la superficie metálica de la plata es el
fundamento principal de su actividad catalítica. Primero, el oxígeno se absorbe
molecularmente a la plata y de esta forma reacciona con el etileno para dar
óxido de etileno. El oxígeno atómico
que se produce no puede formar más óxido de etileno, sino que quema el
etileno y el óxido de etileno, dando CO y H2O:

![]()

Los
catalizadores industriales contienen, en general, hasta un 15% en peso de Ag.
en forma de capa fina depositada en un soporte. Todas las demás características
específicas pertenecen a la pericia de las empresas, que determinan en conjunto
las diferencias tecnológicas en que se
basan los distintos procesos. La
actividad y el comportamiento selectivo de los catalizadores están influidos
principalmente por el método de preparación, la clase de soporte y sus
propiedades físicas, así como por posibles promotores o activadores.
En todos los
procesos se utilizan inhibidores para prevenir la oxidación total. Para ello,
se emplea principalmente 1,2-dicloroetano, que se añade a la mezcla de reacción
en cantidades de unas ppm. El cloro atómico, procedente de la oxidación del HCI
de la deshidrocloración del 1,2-dicloroetano, impide la disociación atómica del
oxígeno y con ello la reacción de combustión en CO2 y H2O.
4.1.2.2.
Realización del proceso
Una de las
principales tareas que se presentan en las instalaciones industriales grandes
es encontrar la forma eficaz de eliminar el considerable calor del conjunto de
reacciones. El recalentamiento del catalizador modifica la distribución óptima
de la plata sobre la superficie del soporte, disminuyendo así la actividad y el
período de vida del catalizador. El mantenimiento de la temperatura adecuada en
el reactor se facilita también con otra medida; la conversión de etileno se
limita, por lo general, a menos del 10% con lo cual es posible limitar el
desprendimiento máximo de calor. Por otra parte, el intenso estudio de
perfeccionamiento de los catalizadores ha conducido a tipos de mayor carga y
más resistentes. Así, por ejemplo, los catalizadores de plata de la Shell en
muchas instalaciones, incluso después de muchos años de trabajo continuado,
muestran solamente una pequeña disminución de la selectividad y rendimiento de
volumen-tiempo. El reactor para óxido de etileno actualmente empleado, contiene el catalizador
rígidamente ordenado en haces tubulares. Con varios miles de tubos, por los que
circula la mezcla de reacción. Como medio calefactor circula entre los tubos un
líquido hirviente, como, por ejemplo, queroseno o tetralina. El calor de
reacción se usa generalmente para producir vapor de, presión media.
Hasta ahora,
los procesos con lechos fluidos (como el Vulcan Atlantic o el de Scientific
Design), como posible solución para una buena eliminación de calor, no han
conseguido ninguna importancia en la industria. Una de las razones principales
es la inadecuada vida del catalizador y
su poca selectividad.
Las primeras
grandes instalaciones industriales, como las de UCC y la Scientific Design en
EE.UU., Distillers en Inglaterra y la IG en la fábrica de Ludwigshafen en
Alemania, usaron aire como medio oxidante. La parte de N2 recarga,
sin embargo, la circulación de gases y da lugar a pérdidas de etileno, ya sea
al eliminar el nitrógeno después del
primer paso de la mezcla de etileno/aire por el reactor, o porque al tener que
usar un segundo reactor, en el que se emplea condiciones de temperatura más
enérgicas, se producen reacciones secundarias con menor selectividad en óxido
de etileno.
Por ello, en
las nuevas instalaciones se usa casi de forma exclusiva el oxígeno como oxidante. A pesar de sus
mayores gastos de inversión y de funcionamiento, por tener que disponer de una
plata para el fraccionamiento del aire, los costos de obtención de óxido de
etileno en total son menores que cuando se emplea aire. De todas formas, en los
procesos de oxidación con oxigeno hay constantemente un 50% de gas inerte, como
metano, etano o CO2, que se usa como gas frenador (absorbente de radicales libres) en ciclo
cerrado. La Shell tiene un funcionamiento este proceso desde mediados de los
años cincuenta. Tiene la ventaja de un menor desprendimiento de gases, pues
sólo alcanza el 2% del desprendimiento de gases del proceso con aire. De esta
forma también se diminuyen considerablemente las pérdidas de etileno. El 60% de
la producción mundial de oxido de etileno el año 1975 fue por el proceso con
oxígeno.
Otra
diferencia respecto a los procesos antiguos es la separación del CO2.
En el proceso con aire se elimina con los gases expulsados, mientras que en el
proceso con O2 se extrae por lavado con disolución de hidróxido
potásico caliente.
Entre tanto,
otras empresas, como la Scientific Design, emplean en sus procesos unas veces
oxígeno y otras aire. La Nippon Shokubai y la SNAM Progetti utilizan otros
procesos industriales.
En la
República Fderal Alemana producen óxido de etileno la BASF, Dow, EC-Dormagen,
Hüls y Hoechst. Las condiciones características de los procesos con O2
son 10-20 bars y 250-300ºC. El contenido de oxígeno de la mezcla de reacción se
fija entre el 6-8% en vol. (Etileno 20-30% en vol.), y con ello fuera de los
límites de explotación de las mezclas de etileno/O2. La selectividad
en óxido de etileno alcanza el 65-70% para una transformación de etileno de
aproximadamente el 8-10%.
Para su
elaboración posterior los gases de reacción se lavan con agua en una torre de
absorción, en la que se disuelve el óxido de etileno. Después se lleva a una
columna de separación (liberador) en que por medio de vapor se separa del agua
y lleva a las columnas de destilación en las que se fracciona. Parcialmente,
las mezclas óxido de etileno-agua se transforman también directamente en
glicol.
4.1.2.3.
Posibilidades de desarrollo de la obtención de óxido de etileno
La economía de
la obtención de óxido de etileno está determinada, sobre todo, por el precio
del etileno. La oxidación total aproximada de 30 mol % de etileno convertido,
constituye uno de los factores básicos del coste. Antes de la crisis del
petróleo de otoño de 1973 la participación del etileno en el coste total era
del 60-70% y en 1974 aumentó al 70-80%.
Es por ello
que son necesarios perfeccionamientos en la tecnología, sobre todo en la
selectividad de los catalizadores, para compensar los costes de la materia
prima.
Las
investigaciones y ensayos para elevar la selectividad merecen por ello una
atención especial, puesto que pueden actuar favorablemente bajo dos puntos de
vista: primero, para alcanzar, efectivamente, un alto rendimiento en óxido de
etileno, con lo cual, al disminuir la oxidación total, además se tendría una
menor tonalidad térmica. Esto, a su vez, permitiría elevar sin peligro la
conversión de etileno, lo que llevaría a una mayor capacidad de producción de
la instalación.
Dos ejemplos
nos pueden mostrar las posibilidades de perfeccionamiento de los catalizadores
con el resultado de aumento de la selectividad:
Así han
conseguido D. Bryce-Smith y colaboradores el desarrollo de un nuevo catalizador
de plata, el llamado <<Clustersilver>>, que se caracteriza por una ordenación
cristalográfica especial de enlaces metal-metal. Se obtiene por descomposición
térmica de cetenuro de plata que se obtiene de la reacción del acetato de plata
con el anhídrido acético. Con dicho catalizador deben ser posibles
transformaciones de etileno del 50-60% con una elevación de selectividad hasta
alcanzar más del 80% en óxido de etileno. De todos modos, aún no se ha
efectuado la prueba en condiciones industriales.
Con el mismo
propósito se ha propuesto el uso de cesio como cocatalizador de la plata. La
selectividad se elevaría con ello al 81%.
Sin embargo,
como del 54 al 67% de la producción mundial de óxido de etileno se destina a la
obtención de glicol, además de los perfeccionamientos en los procesos de
obtención de óxido de etileno, son especialmente interesantes y de un gran
porvenir los de obtención de glicol que no tengan necesariamente que pasar por el relativamente caro óxido de etileno
como producto intermedio
4.2. Productos
derivados del óxido de etileno
El óxido de
etileno, como tal, es tan sólo de aplicación limitada, por ejemplo, como
insecticida en silos de cereales, como esterilizante y como inhibidor de la
fermentación.
Su destacada
importancia reside en la reactividad del anillo oxirano, que lo transforma en
sustancia clave para la obtención de múltiples productos intermedios y
acabados.
El principio
de las reacciones secundarias del óxido de etileno se basa en la apertura
exotérmica del anillo de tres miembros por reactivos nucleófilos, como agua,
alcoholes, amoniaco, aminas, ácidos carboxílicos, fenoles o mercaptanos con
formación de productos etoxilados. En general, cuando reacciona con un
compuesto orgánico, hace que aumente su solubilidad en agua por la adición del
grupo hidroxietilo. La velocidad de reacción se puede aumentar catalíticamente,
tanto por ácidos como por bases. Siempre que no se trabaje a alta temperatura y
presión sin catalizadores, se prefieren los catalizadores ácidos, como ácidos
minerales o cambiadores ácidos de iones.
Puesto que el
producto primario de adición posee un grupo hidróxilo reactivo, puede
adicionarse de nuevo al óxido de etileno. Se forman así progresivamente di-tri
y polietoxilados.
Si se prefiere
obtener el producto de monoetoxilación, se debe emplear óxido de etileno por
defecto.
En la tabla
siguiente se indican los reactivos más importantes que se unen al óxido de
etileno, sus productos de reacción más importantes y otros productos derivados:
|
Reactivos |
Productos de reacción |
Productos derivados |
|
Agua |
Etilenglicol |
Glioxal,
Dioxolano |
|
|
Dietilenglicol |
Dioxano |
|
|
Polietilenglicoles |
|
|
Alcohilfenoles |
Polietoxilatos |
|
|
Alcoholes
grasos |
|
|
|
Äcidos
grasos |
|
|
|
Aminas
grasas |
|
|
|
Amoniaco |
|
|
|
|
Monoetanolamina |
Etilenimina |
|
|
Dietanolamina |
Morfolina |
|
|
Trietanolamina |
|
|
Alcoholes RCH2OH |
Glicolmonoalcohileter |
Glicoldialcohileter |
|
R = H, CH3, n-C3H7 |
Diglicolmonoalcohileter |
Esteres de
glicolmonoalcohileter |
Tabla 2. Productos derivados del óxido de
etileno.
La importancia
relativa de los productos derivados del óxido de etileno se puede deducir
claramente por las prospecciones de uso de óxido de etileno en EE.UU., Japón,
Europa occidental y Alemania occidental.
|
|
EE.UU. 1976 |
1977 |
19781) |
Japón 1977 |
Europa 1976 |
occ. 1977 |
1978 |
Alemania 1976 |
1977 |
1978 |
|
Etilenglicol2) |
50 |
59 |
59 |
71 |
48 |
45 |
49 |
34 |
33 |
36 |
|
Sustancias
tensoactivas no iónicas |
14 |
14 |
13 |
|
18 |
19 |
17 |
26 |
25 |
24 |
|
Etanolaminas |
6 |
6 |
6 |
29 |
8 |
9 |
8 |
14 |
16 |
15 |
|
Éteres
glicólicos |
7 |
7 |
7 |
|
9 |
10 |
9 |
12 |
12 |
12 |
|
Para otras
aplicaciones |
23 |
14 |
15 |
|
17 |
17 |
17 |
14 |
14 |
13 |
1)
Estimado. 2) Incluyendo, en
general, etilenglicoles superiores.
Tabla 3. Empleo del óxido de etileno en
EE.UU., Japón, Europa occ. y Alemania occ. (En %).
4.2.1.
Etilenglicol y etilenglicoles superiores
El
etilenglicol -generalmente designado simplemente glicol- es el producto derivado del óxido de etileno más
importante. En la tabla al margen se resumen las producciones de etilenglicol
de los países industriales más importantes.
El etilenglicol
se obtiene por adición de agua al óxido de etileno:
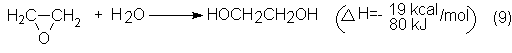
En el proceso
industrial, el óxido de etileno se hace reaccionar con unas diez veces en
exceso molar de agua en fase líquida a presión normal y 50-70ºC en presencia de
un catalizador ácido (por ejemplo,
0,5-1,0% H2SO4), o a 140 hasta 230ºC y 20-40 bars
sin catalizador. La obtención de etilenglicol tiene lugar casi exclusivamente
en un reactor acoplado a la oxidación directa del etileno. La disolución acuosa
resultante de glicol bruto se encuentra por evaporación hasta próximo el 70% y
se fracciona por destilación en varias columnas de destilación al vació.
A pesar del
gran exceso de agua la selectividad en monoetilenglicol es solamente de un 90%.
Al propio tiempo se obtiene un 9% de diglicol, un 1% de triglicol y
etilenglicoles superiores. El rendimiento total llega al 95-96%. Las exigencias
de pureza varían según el empleo a que se destine el glicol; para obtención de
poliésteres se proporciona de calidad especialmente alta (pureza del 99,9% en peso).
Si en la
hidratación del óxido de etileno se disminuye la parte de agua, se forman
progresivamente di, tri y polietilenglicoles:

Otra forma de
obtención parte de la aportación directa de óxido de etileno a los
etilenglicoles. Ordinariamente se realiza a 120-150ºC con una ligera
sobrepresión, frecuentemente en presencia de un catalizador alcalino. Al
aumentar el peso molecular los polietilenglicoles se convierten en líquidos viscosos y, finalmente, en productos
cerosos, pero continúan siendo solubles en agua.
4.2.1.1.
Posibilidades de desarrollo de la obtención de etilenglicol
Los
perfeccionamientos futuros para la obtención de etilenglicol tendrán que ocuparse de mejorar la oxidación
del etileno y la hidratación del óxido de etileno. En primer lugar, están los
trabajos de aumento de la selectividad en ambas etapas de reacción y la disminución del gasto de energía
necesario para el aislamiento del glicol de las disoluciones acuosas diluidas.
En el futuro,
además, deben ganar importancia los procesos de hidratación oxidante directa
del etileno eludiendo la formación del óxido de etileno. De todas formas los
procesos de transformación del etileno en mono y diacetato de glicol (por
ejemplo, Halcon, Tejin, Kuraray, Celanese, DuPont, ICI), a pesar de su muy alta
selectividad de hasta 98%, conlleva la desventaja de las dos etapas, por la
saponificación del acetato y la recuperación del acético:


Halcon como
primera empresa, junto con la Atlantic Richfield (oxirano), según este
principio, ha puesto en marcha en 1978 una planta de etilenglicol, tras superar
las dificultades iniciales, con una capacidad prevista de 360 000 toneladas al
año. La transformación de etileno para dar una mezcla de monoacetato de glicol,
así como principalmente de diacetato, se realiza en acético a unos 170ºC y 28 bars, en presencia de un catalizador
homogéneo que contiene teluro y bromuro. En una transformación de etileno del
60%, se alcanza una selectividad del 96% en acetato de glicol. La hidrólisis
del acetato con H2O, en presencia de un cambiador ácido de iones a
90ºC, alcanza un rendimiento del 95%.
Si en el
futuro también los procesos de oxidación en una sola etapa con catalizadores,
como, por ejemplo, TiO2/HCI (Teijin), CuBr2/CuBr/HBr
(Teijin), combinación I2/Cu (Halcon) o Pd(NO3)2
(Kuraray), llegan a procesos industriales, se alcanzará posiblemente el óptimo
camino para la obtención de glicol a partir de etileno.
El que el
etileno continúe siendo un producto de partida adecuado, si sigue
encareciéndose la nafta, que es su materia prima, podría depender de un
interesante desarrollo de la UCC. Como producto de partida la UCC usa el gas de
síntesis, que seguramente medio o largo plazo representará el producto básico
más barato obtenido por gasificación del carbón. En una reacción a elevada
presión de 1400-3400 bars y a 125-350ºC, el gas de síntesis, en presencia de
rodiocarbonilos complejos, se transforma directamente en glicol, 1,2-propandiol
y glicerina con una selectividad del conjunto del 64%. Como productos
secundarios se forman metanol, formiato de metilo y agua.
4.2.1.2.
Aplicaciones del etilenglicol
El
etilenglicol tiene dos campos de
aplicación principales: como anticongelante del circuito de refrigeración de los motores y como diol para la obtención
de poliésteres. El producto más importante, el tereftalato de polietileno (PET), se emplea principalmente para la
fabricación de fibras, aunque también para láminas y resinas. La
aplicación del etilenglicol en estos
dos campos depende del país y es muy distinta.
En los EE.UU., desde hace tiempo, más del 50% es para el sector de los
anticongelantes (véase la tabla del margen). A causa de la tendencia hacia los
motores pequeños y a la mayor duración de los intervalos entre cambio de
anticongelante, disminuye la proporción hacia estos productos y se espera que
en 1980 se igualarán los consumos de etilenglicol para
anticongelantes y para poliésteres.
En Europa
occidental, en 1977, el reparto del consumo de etilenglicol era de sólo el 36%
para anticongelantes y el 47% para poliésteres; en el Japón, por el contrario,
sólo es el 12% para anticongelantes y, en cambio, el 71% para poliésteres.
Los
polietilenglicoles (etilenglicoles superiores) se usan solos o esterificados,
dependiendo de su peso molecular, como líquidos para frenos, plastificantes o
lubrificantes.
Además, se
usan para la obtención de poliuretanos y de resinas poliéster (ver sección
14.4).
Una pequeña
parte del etilenglicol se destina a productos derivados, como se describirá en
el próximo sección.
4.2.1.3.
Productos derivados: glioxal, dioxolano, 1,4-dioxano
De los
productos derivados del etilenglicol, los más importantes para la industria
son: el glioxal, dioxolano (1,3-dioxaciclopentano) y 1,4-dioxano.
El glioxal se obtiene del glicol por
oxidación en fase gaseosa con aire a unos 300ºC en presencia de catalizadores
de Ag o de Cu, con adición de pequeñas cantidades de combinaciones halogenadas,
como inhibidores de la oxidación total, con rendimientos de hasta el 70%:
![]()
Otra
posibilidad de obtención es la oxidación de acetaldehído con ácido nítrico sin catalizadores o también en presencia de
sales metálicas, como catalizadores. Se realiza con nítrico del 60% bajo
condiciones cuidadosas y 40ºC. Como productos
secundarios se forman también ácido glicólico, oxálico, acético y
fórmico y también como un 10% de ácido glioxílico. Si se aumenta la temperatura
y la concentración de ácido nítrico, se
favorece la oxidación hacia la formación de ácido glioxílico, que sirve para la
obtención de vainilla, etilvanillina y alantoína en cantidades industriales.
La disolución
acuosa de glioxal se purifica por medio de cambiadores de iones. El glioxal
puro es inestable. Por ello se usa en forma de disolución acuosa al 30-40% o
como hidrato sólido con un contenido de glioxal del 80%.
El glioxal se
emplea a causa de la reaccionabilidad de sus dos grupos aldehído frente a
combinaciones polifuncionales con grupos hidróxilos o amino, especialmente para
reacciones de condensación y de reticulación, como, por ejemplo, con urea o sus
derivados, con almidón, celulosa algodón, caseína o cola animal, así como para
acabados textiles y del papel.
Dioxolano
(1,3-dioxaciclopentano) es otro de los derivados del glicol empleado
industrialmente. Se puede obtener por reacción del glicol con formaldehído,
catalizada por protones en medio acuoso:

De la mezcla
de reacción se destila el azeotropo dioxolano/agua, y por extracción, por
ejemplo, con cloruro de metileno, se aísla el dioxolano de la fase acuosa.
El dioxolano
encuentra aplicación análoga a la del óxido de etileno como comonómero en la
polimerización del trioxano a copolímeros del polioximetileno.
El dioxolano
posee un poder disolvente considerable, semejante al del tetrahidrofurano. Como
compuesto acetálico muestra una menor tendencia a la formación de peróxidos que
el éter cíclico, pero frente a esta ventaja tiene la labilidad del enlace
acetálico en medio ácido acuoso.
1,4-Dioxano se obtiene por deshidratación de
glicol o diglicol:

Para ello se
hace actuar sobre el producto empleado ácido sulfúrico diluido u otro ácido
fuerte a 150-160ºC con destilación simultánea del dioxano formado. Otros dos procesos distintos hacen reaccionar clorhidrina etilénica o
2,2-diclorodietiléter con NaOH, con eliminación de CIH y ciclización.
El 1,4-dioxano
se puede también obtener por calentamiento de óxido de etileno con un poco de
sulfúrico o fosfórico concentrados:

El dioxano es
un valioso disolvente para los ésteres de celulosa y éteres de celulosa, así
como también de aceites y resinas. Como éter cíclico forma sales de oxonio y
complejos, por ejemplo, con Br2 o con SO3, que tienen
mucho interés en química preparativa.
4.2.2.
Polietoxilatos
Es la serie de
productos derivados del óxido de etileno, ocupan el segundo puesto los
productos polietoxilados de los alcohilfenoles, alcoholes grasos, ácidos grasos
y aminas alifáticas, por la cantidad que de ellos se obtienen. Por transformación con 10 a 30 moles de
óxido de etileno, los productos de partida pierden su carácter hidrófobo y se
transforman en productos de gran aplicación industrial, cuya hidrófila es
función del número de unidades de óxido de etileno que contiene. Una
solubilidad en agua insuficiente por contener menos de cinco unidades de óxido
de etileno puede mejorarse por esterificación con sulfúrico del hidróxido
final, obteniéndose los denominados éteres sulfatos.
La adición del
óxido de etileno tiene lugar industrialmente, por lo general, a presión de
algunos bars y en presencia de catalizadores básicos como el NaOAc o NaOH a 120-220ºC.
Por lo general, la etoxilación se realiza discontinuamente en calderas con
agitación o en aparatos de circulación cíclica.
Los productos
de etoxilación han encontrado empleo como sustancias tensoactivas no iónicas,
con poca formación de espuma, como
detergentes y humectantes, así como emulgentes y dispersantes.
Así en 1974 se
utilizaron unas 182 000 toneladas de alcoholes lineales etoxilados en la
industria y para uso doméstico en los EE.UU.
La valoración
de estos productos, desde el punto de vista de su amplio empleo, ha sido
influida por el importante criterio que reside en su biodegradabilidad. Es bien
conocido que los alcohilfenoletoxilatos
son biológicamente resistentes, mientras que los etoxilatos de alcoholes
grasos se degradan biológicamente con mayor facilidad. Pero hay que hacer notar
que aun cuando la porción hidrófoba del etoxilato de alcohol graso se degrada
bien, en cambio la parte hidrófila, el poliéster, cuando aumenta la longitud de
la cadena, resulta más difícil degradarlo.
4.2.3.
Etanolaminas y derivados
El óxido de
etileno reacciona exotérmicamente con amoniaco en disolución acuosa al 20-30%,
a 60-150ºC y 30-150 bars, con una gran selectividad, formando una mezcla de las
tres etanolaminas teóricamente posibles:

A presión y a
unos 100ºC, en presencia de una sal de trietanolamina se puede llegar hasta la base cuaternaria. La
composición del producto de reacción puede variarse por diferentes proporciones
de amoniaco y óxido de etileno. Cuanto mayor es el exceso de amoniaco, tanto
más alto es el contenido en monoetanolamina:
|
Proporción
molar |
|
Proporción
de |
selectividad |
|
NH3 : OE |
Mono- |
Di- |
Trietanolamina |
|
10:1 |
75 |
21 |
4 |
|
1:1 |
12 |
23 |
65 |
Tabla 4. Etanolamina a partir de NH3
y óxido de etileno a 30-40ºC y 1,5 bars.
La elevada
proporción de trietanolamina cuando se emplean cantidades equimoleculares de
los reactivos, pone de manifiesto que la reacción primaria con NH3 es más lenta que las reacciones
sucesivas siguientes: Como productos secundarios pueden resultar etoxilatos por
la reacción del óxido de etileno con los grupos OH de la trietanolamina.
Las
etanolaminas son productos industriales valiosos que sirven, sobre todo, para
la obtención de detergentes por reacción de ácidos grasos a 140-160ºC con el
grupo amino o el hidróxilo para dar
amidas etanólicas o ésteres aminados o
por reacción por ambos lados también ésteres amídicos de ácidos grasos en
proporciones variables.
Además, las
etanolaminas se pueden emplear
directamente como bases débiles para la purificación industrial de gases por
separación de gases ácidos, como el SH2 y el CO2. A causa
de su suave reacción básica, las etanolaminas son muy empleadas en la
fabricación de cosméticos, como constituyentes de jabones y cremas. Al
contrario que en los detergentes aquí sólo están como sales de los ácidos
grasos. También sirven, además, para síntesis orgánicas de heterociclos. Las
cifras de producción de los países industriales más importantes -hasta ahora
conocidas -se encuentran reunidas en la tabla del margen.
A partir de
dietanolamina, por deshidratación con sulfúrico del 70% y ciclización, se
obtiene morfolina, que es un disolvente y un producto intermedio:

La etilenimina
es otro producto intermedio importante en la industria, que se puede obtener de
la monoetanolamina. Los procesos industriales actualmente aplicados para su
obtención, generalmente son en dos etapas. Primero se esterifica la etanolamina
con sulfúrico del 95% a b-aminoetilsulfúrico:
![]()
El semiéster
se disocia en la segunda etapa por calentamiento con cantidades
estequiométricas de NaOH a 220-250ºC y 50-80 bars, con lo que se produce la
ciclación a imina y se forma sulfato sódico:

Si la
formación de imina tiene lugar por flujo a través de un tubo de reacción, se
puede regular el tiempo de permanencia en el mismo a 4-10 segundos, con lo cual
se pueden evitar las reacciones secundarias, como, por ejemplo, la formación de
polímeros de la etilenimina. La selectividad en etilenimina alcanza entonces el
80-85% (etanolamina). Procesos de obtención de etilenimina semejantes al
descrito han sido aplicados por la Hoechst y la BASF. Por un proceso de la Dow
la etilenimina se obtiene por reacción de 1,2-dicloroetano con NH3
en presencia de CaO, aproximadamente a 100ºC:

En la
obtención, almacenamiento y transformación de la etilenimina, deben tenerse en
cuenta su alta toxicidad y su fácil tendencia a polimerizar, que en presencia
de pequeñas cantidades de sustancias ácidas puede ocurrir espontáneamente.
La etilenimina
se transforma principalmente en polietilenimina que se emplea en la industria
del papel como sustancia auxiliar. Sirve también como producto intermedio para
síntesis, como, por ejemplo, por reacción con isocianatos, que dan lugar a
ureas.
4.2.4.
Éteres glicólicos
El óxido de
etileno tiene otra importante aplicación por su reacción con alcoholes para dar
monoalcohiléteres del glicol:

Los alcoholes
más empleados en esta reacción son metanol, etanol y n-butanol. El proceso
industrial es semejante a la hidratación del óxido de etileno, con la
diferencia de que generalmente se usan catalizadores básicos, tales como
hidróxidos alcalinos o sus correspondientes alcoholatos o también AI2O3.
A pesar de emplear un gran exceso de alcohol se producen, en general,
reacciones secundarias que dan lugar a di-, tri- y
polietilenglicolmonoalcohiléteres. El etilenglicol monoetiléter se obtiene, por
ejemplo, a 170-190ºC y unos 10-15 bars.
Los
monoalcohiléteres del etilenglicol se emplean con distintos nombres
comerciales, como Cellosolveâ, Carbitolây Dowanolâ, en muchos
campos de actividad, como, por ejemplo, disolventes de pinturas, como
componentes de los líquidos de frenos que determinan su viscosidad, como
emulgentes para aceites minerales y vegetales, así como para la preparación de
pasta para bolígrafos y tintas de imprenta. Las cifras de producción de éteres
de glicol en Alemania occidental y Japón -hasta ahora conocidas- se reúnen en
la tabla del margen. Las cifras para EE.UU. corresponden a
etilenglicolmonoetiléter solamente.
Por
eterificación o esterificación del segundo grupo OH libre en los
monoglicoléteres se obtienen otros disolventes industriales muy importantes,
inertes, apróticos. De estos los más importantes son los dimetilglicoles, como
el dimetilglicol (Glymeâ) y el dimetildiglicol (Diglymeâ).
Los
dialcohiléteres del etilenglicol, tipificados por el dimetiléter, se obtienen
en su mayoría por procesos en dos etapas. En la primera, el monoalcohiléter se
transforma con NaOH y eliminación del agua formada en alcoholato:
![]()
En la segunda
etapa se eterifica el alcoholato sódico con cloruro de alcohilo, generalmente
con CH3CI o con dimetilsulfato:
![]()
En lugar de
este método de obtención con cantidades estequiométricas de
cloro-alcohilderivado e hidróxido sódico, que es tedioso y caro, se ha encontrado
otro mejor para la obtención de etilenglicoldimetiléter. Según éste, se hace
reaccionar primero el monometiléter con formaldehído catalizado por protones
para dar acetales del formol, que después por hidrogenólisis, con un
catalizador especial de Ni, produce cantidades equimoleculares de monometiléter
y dimetiléter, de los correspondientes mono-, di-, tri- o etilenglicoles
superiores:


El
monometiléter resultante entra de nuevo a producir el acetal del formaldehído,
de forma que resulta la ecuación suma siguiente:
![]()
Los
etilenglicoldialcohiléteres se emplean como disolventes especiales muy inertes
y apróticos para reacciones con derivados organometálicos, como, por ejemplo,
reacciones de Grignard o en el dominio de la química del boro, así como para la
fabricación de pinturas de poliuretano. Mezclas de
polietilenglicoldimetiléteres, CH3(OCH2CH2)nOCH3,
con unas cuatro hasta siete unidades de oxietilenos, se emplean como medios de
absorción en el <<Proceso Selexol>> de la Allied. El gas natural o el de
síntesis, por lavado a presión, se liberan de componentes tales como SH2,
CO2 o SO2. Los gases ácidos se liberan finalmente por
expansión fraccionada unos después de otros.
Para la
esterificación de los grupos OH libres, los etilenglicolmonoalcohiléteres se
tratan con ácidos carboxílicos, en especial acético con un catalizador de
protones:

El acetato de
etilenglicol es un extraordinario disolvente para la nitrocelulosa y éteres de
celulosa que se emplea en pinturas y acabados. En 1972 se consumieron en
Alemania Federal 5000 toneladas de acetato de etilglicol.
4.2.5.
Carbonato de 1,2-glicol
El carbonato
de 1,2-glicol -también llamado carbonato de etileno-, originariamente se obtuvo
solamente con glicol fósgeno con eliminación de dos moléculas de HCI.
En 1943 la IG
encontró una nueva forma de obtenerlo a partir de óxido de etileno y CO2:

Hüls ha
perfeccionado por completo este proceso para su aplicación industrial y ha
puesto en marcha una pequeña instalación de producción en Alemania Federal. Con
el mismo proceso funcionan dos plantas en Rumania con una capacidad conjunta de
8000 toneladas/año. Como catalizadores
se emplean aminas terciarias, sales de amonio cuaternarias o carbón activo
impregnado de NaOH. La transformación tiene lugar a 160-200ºC y 70-100 bars.
Los rendimientos alcanzan el 97-98%.
El carbonato
de glicol es un extraordinario disolvente para muchos polímeros y resinas.
Técnicamente se ha hecho uso de ello, por ejemplo, en Rumania para la obtención
de fibras de poliacrilnitrilo. Se usa, además, como producto intermedio para
síntesis orgánicas. Así , por ejemplo, por reacción con amoniaco o con aminas
se pueden obtener carbamatos. El
carbonato de glicol se puede utilizar en casos especiales en lugar del
óxido de etileno para etoxilación.
4.3.
Acetaldehído
Las materias
primas para la obtención del acetaldehído dependen de los países, con
influencia principal de factores económicos e históricos.
En EE.UU.,
Gran Bretaña y Francia, todavía hasta mediados de los años sesenta, desempeñaron
un gran papel los procesos de obtención que parten de etileno, con etanol como
intermedio. En Alemania Federal e Italia, en cambio, se prefería la hidratación
del acetileno por el elevado precio del etanol debido a las medidas fiscales.
La oxidación
de alcanos C3/C4 constituye otros posibles acceso al
acetaldehído, que hasta ahora sólo ha utilizado con una extensión limitada en
los EE.UU. Sólo un 11% de la producción de acetaldehído procedía de la
oxidación de mezclas de propano/butano en 1973.
Con la oferta
de etileno más barato, procedente de disociación de gas natural y nafta, por
una parte, y el desarrollo de procesos industriales de oxidación directa por
Wacker-Hoechst, de otra parte, los antiguos procesos fueron perdiendo
importancia en años sucesivos.
Actualmente,
aún es importante el que procede de
alcoholes EE.UU., Gran Bretaña y Francia; no obstante, su participación en la
producción total de acetaldehído ha descendido marcadamente, llegando a sumar
en 1976 en conjunto sólo 120 000 toneladas, frente a las 600 000 del mundo
entero, es decir, un 17% de la capacidad mundial de acetaldehído.
La obtención
de acetaldehído basándose en acetileno, que en 1968 aún constituía en los
EE.UU. el 7%, se ha suspendido. En el Japón también se cerraron en 1968 las
últimas plantas basadas en acetileno. En cambió, actualmente, en este país el
acetaldehído se obtiene exclusivamente por oxidación de etileno directa. Sin
embargo, aún en 1976, había una
capacidad (mundial) de 264 000 toneladas anuales de acetaldehído repartidas por
todo el mundo (principalmente en los países de Europa oriental). En Alemania
Federal la mayor parte de acetaldehído se obtiene a partir de etileno por el proceso Wacker-Hoechst.
Únicamente la Veba tiene en función una instalación de 80 000 toneladas anuales
para la deshidrogenación de etanol.
Las cifras
conocidas de producción de acetaldehído en los países industriales más
importantes se reúnen en la tabla al margen. A partir de 1974 no se publicaron
las cifras de los Estados Unidos; no obstante, en 1975 la capacidad de
producción ascendía aproximadamente a 730 000 toneladas al año.
4.3.1.
Acetaldehído por oxidación de etileno
4.3.1.1.
Fundamentos químicos
El principio
de la oxidación parcial de etileno para la obtención de acetaldehído, extendida
actualmente en todo el mundo, resulta de la observación hecha por F.C.
Phillips, ya en 1894, de que las sales de platino, en reacción estequiométrica,
oxidaban selectivamente al etileno, dando acetaldehído con simultánea reducción
a platino metálico. Pero sólo el descubrimiento realizado por Wacker de un
proceso catalítico con ayuda de un sistema redox fue decisivo para el
desarrollo de la tecnología adecuada por Wacker Hoechst, que ha permitido su
aplicación industrial en gran escala.
El proceso
total desarrollado por Wacker y Hoechst entre 1957 y 1959, se representa
sencillamente como una oxidación directa catalítica y exotérmica por:
![]()
El catalizador
es un sistema de dos componentes, que consta de PdCI2 y CuCI2.
En el PdCI2 reside la propia función catalítica, que tiene lugar por
la formación de un complejo con el etileno y un cambio de ligandos. Como pasos
parciales más importantes en el mecanismo están la formación de un complejo p (complejo de
transferencia de carga), el reordenamiento a un complejo ó y su disociación a
los productos finales:

El CuCI2
produce la reoxidación del paladio metálico (cerovalente) a su grado de
valencia dos.
Otros
numerosos medios de oxidación se pueden utilizar para transformar el Pdº a Pd2+
, pero sólo el cobre es ventajoso en el sistema redox, ya que por el O2
vuelve a pasar fácilmente de cobre monovalente a cobre divalente por oxidación.
La
anteriormente indicada ecuación global resume un conjunto de reacciones de
varios pasos, que formalmente puede desglosarse en la oxidación rápida de la
olefina:
![]()
y en la
regeneración que determina la velocidad del proceso:
![]()
![]()
La velocidad
de ambas reacciones parciales se puede regular entre las mismas ajustando el
contenido en HCI, viéndose acelerada la regeneración por una concentración
superior de HCI.
En total, las
cantidades de sales de paladio necesarias para la oxidación selectiva del
etileno se puede reducir a cantidades catalíticas empleando un gran exceso de
CuCI2 (ver <<Acetato de vinilo a partir de etileno>>,
sección 9.2.1.2).
4.3.1.2.
Realización del proceso
La obtención
industrial en gran escala de acetaldehído tiene lugar en un sistema de dos
fases, es decir, gas/líquido. Los reactivos gaseosos, etileno, aire o O2
reacciona con la disolución clorhídrica acuosa de los catalizadores en un
reactor de columna de insuflación construido con titanio o revestido de
cerámica.
Se han
desarrollado simultáneamente dos variantes de su funcionamiento:
1. El proceso
en una etapa en el que, en el mismo reactor, se produce simultáneamente la
reacción y regeneración. Como oxidante se emplea O2.
2. El proceso
en dos etapas, en el cual reacción y regeneración se realizan en dos reactores
separados. En este caso se puede utilizar como oxidante aire.
En el proceso
de una etapa se insufla el etileno y O2 a 3 bars y 120-130ºC a
través de la disolución de catalizador. El etileno se transforma en un 35-45%.
El calor desarrollado en la reacción se emplea para destilar el acetaldehído,
así también el agua de la disolución del catalizador, que de nuevo se vuelve al
reactor. De esta forma se introducen en el ciclo unos 2,5-3,0 m3 de
H30 por tonelada de acetaldehído. Para evitar un aumento de gas
inerte, que conduciría a una pérdida de etileno por expulsión, es por lo que es
necesario el empleo de O2 puro y también etileno (del 99,9% en
volumen).
En el proceso
de dos etapas se hace reaccionar el etileno con la disolución catalítica a
105-110ºC y 10 bars hasta casi transformación total. Tras la expansión y
destilación de la mezcla acetaldehído/agua, la disolución catalítica se pasa al
reactor de oxidación y a 100ºC y 10 bars se regenera con aire y, finalmente, se
lleva de nuevo al reactor. Con esto se
produce un consumo de O2 producto del aire empleado y queda así un
gas residual con un elevado contenido de N2 que se puede recuperar
para su empleo como gas inerte. Frente a las ventajas de transformación
completa del etileno empleado y de utilizar aire, se tiene el inconveniente de
inversiones elevadas para el sistema de doble reactor con empleo de altas
presiones y trasvasado de catalizador.
En ambos
procesos el aldehído bruto acuoso se concentra y purifica por una destilación
en dos etapas, que lo liberan de subproductos, como acético, aldehído crotónico
y compuestos clorados. En ambos casos las selectividades son prácticamente
iguales al 94%.
En 1978
existían en todo el mundo instalaciones según
el proceso Wacker-Hoechst, con una capacidad de unos 2,6 millones de
toneladas al año de acetaldehído.
4.3.1.3.
Tendencias de mejoramiento
La mayor parte
(según países entre 50-70%) del acetaldehído se dedica a su transformación en
acético y anhídrido acético, Por tanto, cualquier variación en el mercado del
acético debe reflejarse en la situación del acetaldehído. Si en el futuro ganan importancia otros
procesos más baratos para la obtención de acético, se producirá inevitablemente
un retroceso en la capacidad de
obtención de acetaldehído.
Así, la
carbonilación del metanol, en un sistema general basado en el gas de síntesis, podría ejercer una de esas
presiones económicas (ver sección 4.4.1.3).
También pueden
ser interesantes otros procesos que en lugar del valioso etileno emplean
directamente etano, como, por ejemplo,
con la mezcla fundida de CuO-CuCI2 (Lummus) que lo transforma en acetaldehído,
si se pueden realizar industrialmente y en condiciones económicas.
Otros procesos
posibles, como la oxidación en fase gaseosa del etileno sobre sistemas
catalíticos heterogéneos, como, por ejemplo, a base de Pd-V2O5
(UCC) o la oxidación en fase líquida con catalizadores homogéneos a base de
sales de Pd con heteropoliácidos, como los fosfomolíbdicos o vanádicos (URSS)
no han encontrado hasta ahora ninguna aplicación industrial.
4.3.2.
Acetaldehído a partir de etanol
El
acetaldehído se puede obtener por deshidrogenación catalítica de etanol en
forma análoga a la del formaldehído a partir de metanol:
![]()
Pero al
contrario que para el metanol, no se emplea una oxidación. En lugar de ello,
son corrientes dos modificaciones de la deshidrogenación:
·
Deshidrogenación
sobre catalizadores de plata o preferentemente de cobre, así como
·
Deshidrogenación
oxidante sobre catalizadores de plata en presencia de oxígeno.
Paso 1:
La deshidrogenación de etanol se realiza, por ejemplo, en EE.UU., predominantemente sobre catalizadores de Cu, que están activados con Zn, Co o Cr. Un proceso, utilizado muchas veces, procede de la Carbide and Carbon Corp. La temperatura se mantiene a 270-300ºC, de forma que la conversión del etanol quede limitada al 30-50%. Con ello se consigue una selectividad en acetaldehído del 90%. Como subproductos se obtienen acetato de etilo, alcoholes superiores y etileno. El hidrógeno que se produce simultáneamente se puede utilizar directamente para hidrogenaciones a causa de su pureza.
Paso 2:
Si se realiza la deshidrogenación del etanol en presencia de
aire o 02 (por ejemplo, según el proceso Veba), la combustión
simultánea del hidrógeno formado proporciona el calor necesario para la deshidrogenación (oxideshidrogenación o
deshidrogenación autotérmica):
![]()
En los procesos industriales se prefieren para la
oxidodeshidrogenación catalizadores de plata en forma de redes metálicas o
rellenos cristalinos. Los vapores de
etanol mezclados con aire se dirigen sobre el catalizador a 3 bars y a 450-550ºC. De acuerdo con la cantidad de
aire se establece una temperatura a la cual el calor de oxidación compresa el
calor necesario para la deshidrogenación. Según la temperatura de reacción
resulta una conversión del etanol del 30-50%, por paso, con una selectividad de
un 85-97%. Como subproductos se obtienen acético, fórmico, acetato de etilo, CO
y CO2.
En ambas
modificaciones del proceso se separa el acetaldehído del alcohol sin
transformar y subproductos, y se purifica por diferentes lavados y destilaciones.
El etanol recuperado se emplea de nuevo en la reacción.
4.3.3.
Acetaldehído por oxidación de alcanos C3/C4
Según un
proceso desarrollado por la Celanese, en EE.UU. desde 1943 (capacidad en 1969
unas 160 000 toneladas) se pueden oxidar el propano o las mezclas
propano/butano en fase gaseosa, dando mezclas gaseosas que contienen
acetaldehído. La reacción transcurre radicalariamente sin empleo de
catalizadores a 425-460ºC y 7-20 bars. Una variante de este proceso de
oxidación de butano, de la Celanese, se puede realizar en fase líquida (ver
sección 4.4.1.2). Como oxidante se emplea aire u oxígeno. Alrededor del 15-20%
del hidrocarburo se oxida totalmente. La mezcla de reacción compleja restante
contiene, además de acetaldehído, principalmente formaldehído, metanol.,
acético, n-propanol, metiletilcetona, acetona y otros numerosos productos de
oxidación inevitables.
La
separación de los productos oxidados
tiene lugar, después de la destrucción
de los peróxidos, por medio de variadas y costosas combinaciones de
extracciones y destilaciones.
4.4
Productos derivados del acetaldehído
El
acetaldehído es un importante producto intermedio para la obtención de
numerosos productos orgánicos básicos. Entre ellos se encuentra el acético,
anhídrido acético, cetena-dicetena, acetato de etilo, aldehído crotónico,
n-butanol, 2-etilhexanol, pentaeritrita, cloral, piridinas y muchos más.
La siguiente
tabla de consumos del acetaldehído muestra
claramente que la obtención de acético y su anhídrido consumen la mayor
parte de la producción total de
acetaldehído, a la vez que es la que aumenta más. Además puede comprobarse que
la producción de n-butanol y 2-etilhexanol a partir de acetaldehído se desplaza
progresivamente hacia su producción por hidroformilación de propileno (ver
sección 6.1).
|
|
EE.UU1970 |
1974 |
Japón1970 |
1974 |
Europa occ. 1977 |
Alemania 1975 |
Fed. 1977 |
|
Acético y
anhídrido acético |
45 |
50 |
48 |
58 |
68 |
70 |
68 |
|
n-butanol |
19 |
14 |
18 |
17 |
- |
- |
- |
|
2-etilhexanol |
17 |
11 |
15 |
- |
- |
- |
- |
|
Varios (p.
Ej., acetato de etilo, piridina, alcohilpiridinas y otros) |
19 |
25 |
19 |
25 |
32 |
30 |
32 |
Tabla 5. Consumo de la descomposición del
acetaldehído ( en % en peso)
La porción de
acetaldehído citada como <<Varios>>, dedicada a los productos
especificados, se diferencian mucho según los países; así, por ejemplo, en el
Japón el 14% se dedicó en 1974 al acetato de etilo, mientras que en Alemania
Federal en 1977 la cifra correspondiente llegó al 22%. En los EE.UU. no existe
esta aplicación para el acetaldehído (ver sección 4.4.4). En Japón, en 1974, otra
parte adicional del 7% se dedico a la obtención de ácido paracético (ver
sección 4.4.1.1) y a la cooxidación del p-xileno a ácido tereftálico Estos son
campos de aplicación que casi no tienen importancia en los EE.UU. en la
República Federal Alemania.
4.4.1.
Ácido acético
El ácido
acético es una de las sustancias alifáticas más importantes como producto
intermedio, entre las cuales por su cantidad de producción ocupaba el séptimo
puesto en los EE.UU. en 1978. El
crecimiento de producción es diferente, no obstante, según los países; en el
período de 1960 a 1973 la producción de acético en los EE.UU. y en Alemania
occidental casi se triplicó y, en cambio, en el Japón aumentó siete veces. El
año de recesión, 1975, produjo igual que en casi todos los demás productos de
partida o en los productos intermedios orgánicos, una reducción notable de la
producción que sólo se pudo recuperar en 1976/1977.
|
|
1960 |
1970 |
1973 |
1974 |
1975 |
1976 |
1977 |
1978 |
|
EE.UU. |
336 |
830 |
1100 |
1172 |
997 |
1117 |
1166 |
1264 |
|
RFA |
109 |
238 |
280 |
312 |
225 |
266 |
250 |
271 |
|
Japón |
78 |
401 |
527 |
509 |
443 |
550 |
544 |
n.p. |
n.p. = no
publicado.
Tabla 6. Producción de ácido acético ( en 1000
toneladas).
Las cifras de
producción indicadas se refieren sólo a la síntesis de acético. Además, en
algunos países se obtiene en cantidades limitadas por fermentación de substratos alcohólicos (vinagre de
fermentación), principalmente para uso culinario, y por destilación de madera
(vinagre de madera). Así, por ejemplo, en Alemania Federal se obtuvieron en 1975 unas 10 400 toneladas de vinagre de
fermentación (calculado como acético al 100%). En lo que sigue entenderemos
como ácido acético solamente el acético de síntesis.
El acético de
síntesis, durante mucho tiempo, se obtuvo principalmente a partir de
acetaldehído. Ya al principio de la primera guerra mundial estaban trabajando a
escala industrial los procesos de oxidación de la Hoechst, Wacker y Shawinigan.
Por esto el
proceso de obtención del acético estaba muy ligado al del acetaldehído, lo cual
hizo que experimentasen juntos el desplazamiento de su materia prima del acetileno al etileno.
La necesidad
económica de aprovechar y valorizar los hidrocarburos inferiores llevó, tanto
en los EE.UU., como en Inglaterra y Alemania Federal, a investigar los procesos
de oxidación de parafinas ligeras a la BP, Celanese, British Distillers, Hüls y
UCC, así como de las olefinas C4 a la Bayer y Hüls.
Aunque por los
años veinte comenzaron los primeros intentos de carbonilar el metanol, sólo
bastante más tarde fue la BASF la que
halló la solución industrial. Hace pocos años se llegó a otra solución en este
sentido, por la Monsanto, que con la catálisis con rodio ha adquirido un
impulso nuevo e importante.
4.4.1.1. Ácido acético por oxidación de acetaldehído
La oxidación
del acetaldehído con aire o con O2 a acético transcurre como
reacción de radicales a través del ácido paracético como producto intermedio.
En detalle se
produce primero la iniciación de la reacción por un radical acetilo, que con O2
forma el radical peróxido terminado con la formación del ácido paracético.
Aunque éste puede sufrir una homólisis del grupo peróxido y pasar a acético, se
supone, sin embargo, que el paracético reacciona preferentemente con
acetaldehído, dando un a-hidroxetilparacetato,
que después por un mecanismo de transición cíclico se disocia en dos moléculas
de acético:

Así pues,
también el ácido paracético puede ser el producto principal si la oxidación se
realiza en condiciones suaves y preferentemente sin catalizador y en un
disolvente, como, por ejemplo, acetato de etilo, a -15 hasta 40ºC y 25-40 bars
y con aire.
La UCC y FMC
en EE.UU., la Daicel en Japón y la British Celanese en Inglaterra, han puesto
en funcionamiento plantas industriales en las que se obtiene ácido paracético,
según el principio precedente.
Si se usa un
catalizador redox para la oxidación de acetaldehído a acético, el catalizador
sólo sirve para formar radicales acetilo y, por tanto, para iniciar la reacción
de oxidación, sino también para acelerar el proceso de descomposición del ácido
peracético con formación de un radical acetoxi y ramificación de la cadena.
Como
catalizadores se utilizan principalmente disoluciones de acetatos de Co o de
Mn.
La oxidación
actualmente se realiza sobre todo con oxígeno, por ejemplo, según el proceso de
la Hoechst continuo, a 50-70ºC en torres de oxidación de acero inoxidable
(columnas de soplado) y con acético como disolvente. Por lo cual es necesario
realizar la transformación por lo menos a 50ºC para alcanzar una abundante
descomposición del peróxido y con ello una suficiente velocidad de oxidación.
El calor de la reacción se elimina por circulación de la mezcla de oxidación a través de un sistema de
refrigeración. El cuidadoso control de
la temperatura permite limitar la degradación oxidante del acético a fórmico,
CO2, pequeñas cantidades de CO y H20. La selectividad en
acético alcanza el 95-97% (CH3CHO).
Como variante
de la oxidación de acetaldehído con oxígeno, la Rhone-Poulenc/Melle Bezons ha
desarrollado un proceso que emplea aire
como oxidante. Con él se pueden conseguir selectividades semejantes a las que
da el oxígeno puro. Sin embargo, la desventaja es la gran cantidad de gas
inerte en la oxidación con aire, que contiene las correspondientes presiones
parciales de acetaldehído y acético, que se tienen que eliminar por lavado.
Ambos procesos
de oxidación se semejan además en los subproductos, a los que además del CO2
y fórmico hay que agregar también acetato de metilo, metanol, formiato de
metilo y formaldehído. Se separan por destilación, con lo que resulta acético
anhidro directamente, puesto que los subproductos se comportan, en la
aliminación de agua, como medios de arrastre.
4.4.1.2.
Ácido acético por oxidación de alcanos y alquenos
Para la
obtención de acético por degradación oxidante se prefiere emplear como
productos de partida los hidrocarburos C4-C8. Se pueden
reunir, según los diferentes procesos desarrollados, en los siguientes grupos:
·
n-butano
(Celanese, Hüls, UCC).
·
n-butenos
(Bayer a través de acetato de sec-butilo como producto intermedio, Hüls
directamente).
·
Bencina ligera
(BP, Distillers).
Caso 1:
En 1973, la capacidad de producción de acético en los EE.UU.
se basaba en un 40% en la oxidación en fase líquida de n-butano, como hacen la
Celanese y la UCC. Así la instalación de la UCC en Texas produce unas 225 000
toneladas al año de acético, 36 000 de metiletilcetona y 23 000 de fórmico,
por oxidación sin catalizadores, de n-butano con oxígeno a 15-20 bars y
180ºC, empleado como medio de reacción los productos de oxidación líquidos. La
excesiva degradación oxidante se controla por una limitación de la conversión
al 10-20%. Desde 1965 funciona también
en Holanda una instalación con una capacidad anual de acético de 50 000
toneladas, según el proceso Celanese LPO (oxidación en fase líquida), que entre
tanto ha sido adquirida por la Akzo y ampliada en 1977 a 100 000 toneladas anuales.
El proceso LPO de la Celanese trabaja a 175ºC y 54 bars con
acetato de cobalto como catalizador. Una parte de los numerosos subproductos,
una vez separado el acético, se vuelven
a emplear en el proceso y bien se transforman en acético o bien se
oxidan totalmente. De esta forma se simplifica el proceso. Además del acético
se asilan acetaldehído, acetona, metiletilcetona, acetato de etilo y metanol.
Si es necesario y a costa del rendimiento en acético, se
puede obtener metiletilcetona hasta un 17% de la capacidad de la instalación.
También la Hüls ha mantenido en funcionamiento varios años
una oxidación de n-butano, en este caso sin catalizador, en una planta
industrial con capacidad de unas 20 000 toneladas al año. La oxidación se
realiza a 60-80 bars y a 170-200ºC con aire o con enriqueciendo en O2
(cera del 30% de O2) en fase líquida, formada por acético bruto. La
conversión de n-butano, para evitar reacciones subsiguientes, se limitaba al
2%. Como producto principal se obtenía,
en efecto, acético con una selectividad del 60%, además de numerosos
subproductos, como acetona, metiletilcetona, acetatos de metilo y etilo, y
pequeñas cantidades de ácidos fórmico y propiónico. La elaboración exigía
una instalación de destilación en varias etapas, por ejemplo 14
columnas a presión normal, así como otras con sobrepresión o baja presión.
Caso 2:
Hüls desarrolló aún otro proceso para obtención de acético por oxidación de n-butenos. Este proceso funcionó durante algún tiempo en una instalación de prueba, pero hasta ahora no se ha montado ninguna instalación a escala industrial. En esta forma el n-buteno se oxida con ligera sobrepresión y a 200ºC, en la fase líquida, formada principalmente por el acético bruto resultante. Como catalizadores se usan vanadatos de titanio o de estaño. Por diversas causas, por ejemplo, por los estrechos límites de las mezclas explosivas, la oxidación tiene lugar en presencia de gran cantidad de vapor de agua. Por esto, el acético resultante es muy diluido y es necesario un gasto grande de energía para concentrarlo, hasta un 95% de acético. Con una conversión del 75% de buteno se obtiene una selectividad en ácido bruto del 73%.
Bayer se dirige, con su proceso en fase líquida, a la
obtención de acético a partir de n-butenos en dos etapas por otro camino. De la
fracción de craqueo C4, después de separar el butadieno y el
i-buteno (ver sección 3.3.2), resulta una mezcla de 1-butenos y cis y trans-2-buteno. Esta se transforma
en la forma indicada en la ecuación siguiente, en 2-acetoxibutano, es decir, en
acetato de sec-butilo:

La adición de acético tiene lugar a 100-120ºC y 15 a 25
bars, con cambiadores ácidos de iones que contienen grupos sulfónicos y produce
con simultánea isomerización de los n-butenos, exclusivamente 2-acetoxibutano.
Éste, en una segunda etapa a 200ºC y 60 bars, se oxida con aire a ácido acético en una reacción en fase
líquida no catalizada:

Después de la elaboración de la mezcla de reacción en una
destilación azeotrópica y otra normal, se recicla una parte del acético para la
obtención del acetato de sec-butilo.
La selectividad en acético alcanza el 60% y como
subproductos más importantes se obtienen ácido fórmico y CO2.
Hasta ahora no se ha construido ninguna instalación grande.
Caso 3:
La British Distillers ha desarrollado en Inglaterra un
proceso para la oxidación de destilados brutos con un intervalo de temperatura
de ebullición de 15-95ºC, que corresponde a una bencina ligera, en el campo C4-C8.
La oxidación por aire tiene lugar en fase líquida a 160-200ºC y presión de
40-50 bars en un reactor de acero
inoxidable, sin catalizador, por un mecanismo radicalario. El producto
de oxidación se separa en una destilación en dos etapas, obteniendo producto de
partida y subproductos ligeros y volátiles, así como una mezcla de ácidos
acuosa, de la cual se extraen todos los ácidos por un disolvente de punto de
ebullición bajo, como, por ejemplo, el acetato de i-amilo, que extrae primero
los ácidos y un poco de agua. La fase orgánica se destila luego, separándose en
sus componentes. Además del acético, se forman principalmente fórmico y
propiónico y pequeñas cantidades de succínico. Otro producto que se obtiene en cantidad
tal que merece aislarlo es la acetona.
Todos los subproductos contribuyen a la economía del proceso. Según las
condiciones del mismo, se pueden obtener unas 0,35 a 0,75 toneladas de
subproductos por cada tonelada de acético. La BP ha perfeccionado el proceso de
la Distillers y especialmente en Inglaterra ha instalado varias fábricas muy
bien conseguidas. En 1978 la capacidad llegaba a unas 180 000 toneladas de
acético y unas 18 000 de propiónico. A partir de 1979 la capacidad de las
instalaciones de la BP se amplió en 150 000 toneladas, aunque según el proceso
Monsanto a base de metanol.
4.4.1.3.
Acético por carbonilación de metanol
La BASF se
ocupó en investigaciones para la transformación catalítica de CO y H2
que fueron también preparadoras del
camino a un nuevo proceso de obtención de acético. Hacia 1913 se descubrió que
el metanol, producto primario de la reacción del gas de síntesis, se podía carbonilar a acético. Este camino
interesante a partir de 1920 cuando se dispuso de metanol en cantidades
industriales. A partir de entonces se ocuparon del problema otras empresas, por
ejemplo, la British Celanese a partir de 1925, intensamente en la
carbonilación, que transcurre según la ecuación siguiente:
![]()
Los problemas
de corrosión que desde el principio se plantearon, sólo se pudieron
resolver a finales de los años cincuenta con el empleo de
nuevas aleaciones resistentes de molibdeno-níquel (Hastelloy). En 1960 se puso
a funcionar una pequeña instalación por la BASF. En el proceso industrial (BASF)
el metanol solo o mezclado con dimetiléter y poca agua a 250ºC y 680 bars se
hace reaccionar con CO en presencia de CoI2 en fase líquida. En esta
forma, catión y anión actúan con funciones diferentes en el mecanismo de
reacción; se supone que primeramente el ioduro de cobalto se transforma en
hidruro de cobalto tetracarbonilo y ioduro de hidrógeno, que con el metanol da
ioduro de metilo:
![]()
![]()
El hidruro de
cobalto tetracarbonilo y el ioduro de metilo reaccionan para dar el importante
intermedio CH3Co(CO)4 que por inclusión de CO y posterior
hidrólisis se convierte en acético y se recupera el hidruro de cobalto
tetracarbonilo:
![]()
![]()
Con lo cual,
ambos componentes del catalizador están de nuevo a disposición para repartir la
reacción. En el proceso industrial, tanto el cobalto, como el iodo se pueden
recuperar casi completamente. La selectividad en acético alcanza el 90% (CH3OH)
y el 70% (CO). Por cada 100 kg. de acético se obtienen 4 kg. de subproductos,
que están constituidos por múltiples especies químicas. El CO2 debe
ser considerado como coproducto [según la
ecuación (41)]. Por destilación del producto bruto en
cinco columnas se obtiene acético del
99,8%. En 1976 había en función dos instalaciones según el proceso BASF, una en
la República Federal Alemana con capacidad anual de 35 000 toneladas y otra en
Borden, en EE.UU., con capacidad anual
de 52 000 toneladas.
A mitad de los
años sesenta, encontró la Monsanto que
el rodio combinado con iodo proporcionaba un catalizador mucho más activo para
la carbonilación del metanol que el ioduro de cobalto. Igual que con éste, se
supone que el complejo de rodiocarbonilo que se forma desempeña con el metil
ligando en forma de [CH3-Rh(CO)2I3]- la especie
activa. Por interposición de CO en el enlace CH3-Rh se forma un
complejo de acetil-rodio, que, por ejemplo, por metanólisis, puede volver a
reaccionar para dar acético y el complejo de partida:

En 1970 se
puso en marcha una primera instalación industrial en Texas City con capacidad
anual de 150 000 toneladas de acético.
Posteriormente
se han construido otras instalaciones de acético, con preferencia según el
proceso Monsanto, de modo que, por ejemplo en los EE.UU., ya en 1978 se
obtenían por este proceso el 17% de la producción de ácido acético.
En el proceso
industrial, reaccionan continuamente metanol y CO en la fase líquida a
150-200ºC y a ligera sobrepresión, formando ácido acético con selectividades
del 99% (CH3OH) y superior
al 90% (CO). Los principales subproductos de la reacción de conversión son CO2
y H2. En una planta grande moderna el control del proceso totalmente
automatizado comprende también la producción y regeneración del sistema
catalizador, ya que para la economía del proceso es un factor determinante el reciclado del rodio
con pocas pérdidas.
4.4.1.4.
Posibilidades de desarrollo en la obtención del ácido acético.
La
disponibilidad y evolución de los precios de las materias primas adecuadas para
la obtención de acético, experimentan una constante variación y dificultan la
fiabilidad de un pronóstico. Los cambios que se están produciendo en la química
del gas de síntesis a base de petróleo y de carbón hacen aparecer el horizonte
de la carbonilación del metanol para la obtención de acético como prometedor.
De todas formas, en el proceso de la Monsanto existe el problema central de las
pequeñas perdías en el ciclo del rodio a causa de su elevado precio y su
limitada disponibilidad. Por ello, merecen especial atención las mejoras que se
puedan conseguir en el proceso histórico de carbonilación del metanol a alta
presión. En las innovaciones introducidas por la BASF y la Shell se ha
demostrado que no son necesarias tan elevadas presiones y temperaturas y que,
por ejemplo, a temperaturas de 80-200ºC y 70-300 bars se puede realizar la
carbonilación, si se añaden sales o complejos de platino, al Co/I2
utilizado como catalizador.
La UCC ha
anunciado un nuevo proceso basado únicamente en gas de síntesis para la
obtención de acético, etanol y acetaldehído. Como catalizador se emplea rodio
depositado sobre a-AI2O3 o SiO2 a 150-450ºC y
1-700 bars.
Los procesos
de obtención de acético a base de etileno se distinguen por su alta
selectividad, pero pueden perder su posición preferente si continúa la rápida
elevación de precio del etileno iniciada en 1973 y no transmite con la misma
velocidad de encarecimiento a. Otros productos de pendientes del petróleo para
su fabricación, que sean adecuados para la obtención de acético.
Visto así,
abundan en las refinerías cantidades crecientes de las fracciones C4-C8,
pero también de butano del gas natural que posiblemente son productos de
partida más baratos.
Para la
valoración total de un proceso interesa considerar no sólo los costes de
material, sino también los de fabricación y los de inversión, y, sobre todo, la
selectividad, ya que los productos secundarios tienen también que poderse vender. Esta interdependencia de los
factores mencionados y aspectos coyuntural se puede apreciar en el anuncio de
la Celanese de poner en marcha para 1978 una nueva instalación de acético con
una capacidad anual de 272 000 toneladas, según el proceso de Monsanto, después
que ella misma posee tres instalaciones de acético, de las cuales dos son a
base de etileno-acetaldehído y la otra a base de butano.
4.4.1.5.
Aplicaciones del ácido acético
El acético se
usa, principalmente, para la obtención de diferentes ésteres acéticos, como se
aprecia claramente en las cifras de su consumo en los EE.UU.
|
Producto |
1973 |
1976 |
19801) |
|
Acetato de
vinilo |
40 |
43 |
46 |
|
Acetato de
celulosa |
23 |
22 |
18 |
|
Acetatos de
butilo e i-propilo |
12 |
12 |
12 |
|
Anhídrido
acético, acetanilida, cloruro de acetilo, acetamida |
10 |
10 |
10 |
|
Disolvente
para la obtención de tereftálico y su éster dimetílico |
10 |
10 |
11 |
|
Cloroacético |
3 |
2 |
2 |
|
Otros prod.
diferentes |
2 |
1 |
1 |
1) Previsto.
Tabla 7. Productos derivados del acético en
los EE.UU. (% en peso).
El orden de
importancia entre los ésteres es, en la mayoría de los países, semejante; la
máxima importancia la tiene el acetato de vinilo y le sigue el acetato de
celulosa. Así, en Europa occidental, en 1978, con una producción total de unos
0,86 millones de toneladas de acético se consumieron para acetato de vinilo el
33% y el 7% para acetato de celulosa. En el Japón, con una producción total en
1976 de casi 0,54 millones de toneladas, la parte utilizada para acetato de
vinilo del 25% y para acetato de
celulosa el 11%.
También en la
República Federal Alemana, en 1976, se obtuvieron unas 157 000 toneladas de
acetato de vinilo, que constituyen la
mayor participación en ésteres acéticos. En este caso, sin embargo, sigue en
segundo lugar el acetato de etilo con unas 72 000 toneladas (ver <<Reacción de Tischenko>> ,
sección 4.4.4), y sólo después el acetato de celulosa con 35 000
toneladas. Además se obtienen acetatos de n-butilo e i-butilo, así como acetato
de metilo, ésteres importantes que, como el acetato de etilo, se utilizan
preferentemente como disolventes para pinturas y resinas.
En el acetato
de celulosa, según el grado de acetilación en que se esterifican los tres
grupos OH en cada molécula de glucosa, el contenido de acetato está entre el
5262,5% en peso. Se utiliza para obtener fibras, películas y pinturas.
Las sales de
acético, como por ejemplo, los acetatos de Na, Pb, AI y Zn se utilizan como
sustancias auxiliares en las industrias textil y de cuero, en tintorería y en
medicina.
Los ácidos
cloracéticos son también derivados importantes del acético. De ellos, el monocloracético es el de mayor
importancia. La producción mundial de
monocloracético se estima en unas 180 000 toneladas al año, de las cuales
corresponden a Europa occidental unas 120 000 toneladas. La obtención de
monocloracético tiene lugar por cloración de acético en fase líquida a unos
85ºC, generalmente sin catalizador, pero frecuentemente con adición de
iniciadores como anhídrido acético o cloruro de acetilo.
El
monocloracético es un producto de partida importante para la obtención de
carboximetilcelulosa y un producto intermedio para agentes fitosanitarios,
colorantes y productos farmacéuticos.
El anhídrido
acético y la cetena, productos derivados del acético, de importancia
industrial, que son también productos intermedios, se considerarán en los
capítulos siguientes.
El acético se
usa también como disolvente en las oxidaciones en fase líquida, como, por
ejemplo, la del p-xileno a tereftálico o dimetiltereftalato.
Otro campo de
aplicación del acético es la fermentación para obtener L-lisina o L-glutámico.
En el proceso de Ajimoto o Kyowa Hakko, se emplea acético junto con otros
nutrientes ordinarios, como fuente principal de carbono. Si se emplea acético
en forma de acetato amónico, se aportan el nitrógeno y el carbono a la
fermentación mediante un solo portador. AL contrario que en el proceso de
síntesis en que se obtiene DL-lisina (ver apartados 10.3.1.4 y 11.3.3), los procesos de fermentación
producen directamente L-lisina. Como otros nutrientes para la obtención de
L-lisina se emplean también hidratos de carbono, tales como productos del maíz
o melazas.
4.4.2.
Anhídrido acético y cetena
El anhídrido
acético, intermolecular, y la cetena, intramolecular, son anhídridos del
acético, que en su fabricación, aplicación e importancia son muy semejantes al
ácido acético.
Las cantidades
de producción de anhídrido acético conocidas de los principales países
industriales se indican en la tabla al margen.
La cetena y su
dímero, la dicetena, son cuantitativamente de menor importancia. En 1973, en la
RFA las capacidades ascendían aproximadamente a 20-30 000 toneladas anuales.
El anhídrido
acético se obtiene industrialmente por dos diferentes caminos. El uno es un
proceso de oxidación de acetaldehído modificado; el otro transcurre por la
cetena como producto intermedio que, generalmente, se obtiene por
deshidratación de ácido acético y finalmente se le hace reaccionar con ácido
acético. Otra forma de obtener la cetena procede de la acetona. Así, por
ejemplo, en la RFA en la actualidad, como dos terceras partes de la producción de anhídrido acético son
por oxidación de acetaldehído y la tercera parte restante por acetilación de
acético con cetena.
El proceso
modificado de oxidación de acetaldehído lo desarrolló la Hoechst-Knapsack, así
como con una variante la Shawinigan. En lugar del acetato de manganeso, como
catalizador se emplea una mezcla de acetatos de cobre y cobalto. La reacción es
a unos 50ºC y 3-4bars.
El radical
acetilo producido por sustracción de hidrógeno en el acetaldehído (ver <<Acético por oxidación de acetaldehído>>, sección 4.4.1.1.) se oxida con Cu2+
a acetilcatión, que reacciona a su vez con acético dando anhídrido:

El ácido
peracético se obtiene paralelamente por adición de O2 al radical
acetilo que sustrae hidrógeno del acetaldehído. El perácido sirve para reoxidar
Cu+ a Cu2+:

El acético que
se produce a partir del radical acetóxilo es un coproducto necesario.
El agua
formada puede ahora, a su vez, iniciar la reacción siguiente del anhídrido
acético a ácido acético, sino se elimina rápidamente del equilibrio por
destilación con un medio de arrastre, como, por ejemplo, acetato de etilo en el
proceso industrial. Para una conversión del 95% de acetaldehído se alcanza una
proporción óptima de anhídrido/ácido del 56/44.
En el proceso
de obtención de anhídrido acético por deshidratación de ácido acético a través
de cetena (proceso Wacker), se hace la termólisis del ácido acético en
presencia de fosfato de trietilo a 700-750ºC y a presión reducida, formándose
cetena y agua:
![]()
Finalmente, se
neutraliza el fosfórico producido con NH3 o piridina aun en fase
gaseosa y se enfrían rápidamente los gases de disociación para congelar el
equilibrio.
Las sustancias
de punto de ebullición alto que acompañan ala cetena (anhídrido acético, ácido
acético y agua) se separan de la cetena gaseosa en un sistema de enfriamiento
escalonado. Los productos separados, una vez
eliminada el agua, se reciclan para la disociación. La conversión de
ácido acético es del 80% y la selectividad en cetena superior al 90% (acético.
La cetena así purificada se hace reaccionar directamente con ácido acético
(como en el proceso Wacker) a 45-55ºC, enviándola con una bomba de anillo
líquido, y se convierte en anhídrido acético:
![]()
En esta etapa
se alcanza aproximadamente un 100% de selectividad. Esta obtención de anhídrido
acético posee la ventaja de poder obtener la cetena como producto intermedio
cuando convenga. Además puede emplearse ácido acético del proceso más económico
o el resultante de las reacciones de acetilación con anhídrido acético.
La cetena se
puede obtener también independientemente del ácido acético por termólisis de
acetona a 600-700ºC en presencia de un poco de CS2:
![]()
Para una
conversión de acetona del 25% se puede alcanzar una selectividad en cetena del
70-80%. La termólisis de acetona tiene poca importancia en la industria: en
EE.UU. hay una instalación funcionando en la Hoffman La Roche y también en la
URSS se practica la obtención de cetena
a partir de acetona.
El anhídrido
acético se emplea, sobre todo, como agente acetilante, especialmente para la
obtención de acetilcelulosa, de productos farmacéuticos, como, por ejemplo,
acetilsalicílico, acetanilida entre otros
y de productos intermedios. La cetena se emplea también como agente acetilante. El caso especial de
acilación de acético para dar anhídrido es el más significativo.
Una aplicación
interesante de la cetena son las reacciones de adición, entre las cuales cuenta
la dimerización a dicetena.
La dicetena se
obtiene por dimerización de cetena en torres de flujo descendente, en las
cuales se introduce la cetena a 35-40ºC en contracorriente con la dicetena
líquida:

La transformación
transcurre muy bien; las pequeñas cantidades de cetena en los gases residuales
se eliminan por lavado con acético diluido. La dicetena se obtiene pura por
destilación muy cuidadosa, pues se polimeriza lo mismo en medio alcalino que en
medio ácido.
La dicetena es
un producto de partida importante para la obtención de derivados del
acetilacético, que se obtienen por adición catalizada por ácido o base
alcalina, es decir, por apertura del anillo b-lactónico con
alcoholes, amoniaco, aminas, hidrazinas y otros.
Las
acetilacetanilidas, con sustitución en el anillo fenílico al igual que los
derivados de la pirazolona obtenidos de
la dicetena y fenilhidrazinas sustituidas, tienen gran aplicación como
productos intermedios para colorantes y fármacos.
La cetena
monómera forma b-lactonas con aldehídos, como, por
ejemplo, formaldehído en presencia de
AICI3 que da b-propiolactona:

Como aldehído
crotónico reacciona la cetena para dar una b-lactona lábil
que se puede estabilizar como poliéster
(ver <<Ácido sórbico>> ,
sección 4.4.3).
4.4.3.
Condensación aldólica del acetaldehído y productos derivados
El
actetaldehído, como aldehído con una H en a activo,
puede reaccionar de forma característica para formar el dímero
acetaldol:
![]()
El
acetaldehído se transforma así, análogamente a como se hacía en un viejo
proceso de la IG, reaccionando a 20-25ºC mientras circula por un tubo, con
tiempo de permanencia de algunas horas en presencia de disoluciones diluidas de
NaOH. La conversión de acetaldehído en la reacción de aldolización es de un
50-60% para evitar la formación de resina y de otros subproductos y derivados.
La reacción se
para por adición de ácido acético o fosfórico. El aldol se obtiene en forma de
disolución acuosa de un 73%, después de eliminar por evaporación el
acetaldehído no transformado. La selectividad en aldol alcanza el 85%. Como subproducto sólo se
forma prácticamente aldehído crotónico.
El aldol,
evitando cuidadosamente la fácil deshidratación, se puede hidrogenar a
1,3-butandiol.
La cantidad
sobrante se utiliza, sin embargo, para obtención de crotonaldehído. La
deshidratación transcurre muy fácilmente en presencia de un poco de ácido acético, que simultáneamente impide
otras condensaciones. El agua se elimina por destilación a 90-110ºC:
![]()
El aldehído
crotónico se purifica por una destilación en dos etapas. La selectividad
alcanza el 95% (CH3CHO). La Hoechst posee en Alemania una
instalación para crotonaldehído con capacidad de unas 14-15 000 toneladas al
año. El crotonaldehído era antes un producto de partida importante para la
obtención de n-butiraldehído por hidrogenación parcial y su rehidrogenación a
n-butanol. Ambos productos se obtienen actualmente en la Alemania Federal y,
por ejemplo, en Inglaterra y Francia, exclusivamente, por hidroformilación de
propeno (ver sección 6.1).
En EE.UU. y
Japón se mantiene aún la vía de acetaldehído para la obtención de n-butanol. Se
realiza por hidrogenación del aldehído crotónico con catalizadores de Cu-Cr a
170-180ºC o con catalizadores de níquel
a temperaturas más bajas. Así, por ejemplo, en Japón la proporción de
consumo de acetaldehído para obtener n-butanol seguía siendo en 1975 un 17%.
Otro producto
derivado del n-butiraldehído es el 2-etilhexanol, que se puede obtener por
condensación aldólica y posterior hidrogenación (ver sección 6.1.4.3).
La serie de
reacciones en múltiples etapas, desde acetaldehído a n-butiraldehído y
2-etilhexanol, sólo tiene alguna importancia en EE.UU., donde se obtuvo todavía
en 1970 un 50% del 2-etilhexanol por ese camino. Para 1973 esta proporción en la producción total de 182 000
toneladas de 2EH descendió a menos del
10%.
El aldehído
crotónico tiene además una importancia creciente para la obtención del ácido trans-trans-2,4-hexadinoico, denominado
ácido sórbico. El crotonaldehído reacciona primeramente con cetena en un
disolvente inerte (tolueno, por ejemplo) a 30-60ºC, dando un poliéster. Como
catalizadores se usan sales solubles de Zn o Cd de ácidos carboxílicos
superiores. En la segunda etapa, el poliéster de despolimeriza a ácido sórbico
por termólisis o por hidrólisis catalizada por protones:

El ácido
sórbico libre y sus sales de potasio o calcio tienen gran importancia para la
conservación de alimentos o condimentos. En Alemania lo obtienen la Hoechst. En
EE.UU. la Monsanto en 1977 puso en marcha la nueva producción de ácido sórbico
basada en el proceso aldehído crotónico/cetena, después que la primera y única
instalación industrial de ácido sórbico en EE.UU. de la UCC se parara en 1970.
La base de este proceso era el aldehído sórbico (2,4-hexadienal) que por
oxidación catalizada por plata lo transformaba en ácido sórbico. El propio
aldehído sórbico se obtenía por condensación aldólica de acetaldehído en
presencia de sales de aminas secundarias. En el Japón hay varios fabricantes.
El consumo mundial de ácido sórbico para 1978 se estimó en unas 12 000
toneladas para una capacidad de producción de cerca de 18 000 toneladas al año.
Otro producto
de adición del crotonaldehído se obtiene por reacción con metanol. El
3-metoxibutanal resultante se disocia fácilmente en sus productos de partida,
por lo que hay que hidrogenarlo inmediatamente a 3-metoxibutanol. Para su
obtención se emplea un exceso de
metanol en presencia de hidróxido sódico y temperatura inferior a 5ºC, con lo
que se adiciona el metanol al crotonaldehído y da el 3-metoxibutanal, que, sin
aislar y en fase líquida, se hidrogena
en presencia de catalizadores de Ni o preferentemente de Cu:

La
selectividad en 3-metoxibutanol alcanza un 90% aproximadamente (CH3-CH=CH-CHO).
El
alcohol-éter es un constituyente de líquidos hidráulicos; su acetato se utiliza
como disolvente extraordinario para pinturas.
Otro derivado
del aldehído crotónico es el ácido crotónico. Se obtiene por oxidación en fase
líquida del aldehído con aire o O2 a baja temperatura (20ºC) y a 3-5
bars:
![]()
En la reacción
se forma, predominantemente la forma más estable, el ácido trans-crotónico, con
una selectividad aproximada del 60% (crotonaldehído). Se obtiene en Alemania
por Hoechst, en EE.UU por Eastman Kodak, así como por varios fabricantes en
Japón y se utiliza como componente para copolimerizaciones. Una pequeña parte
se emplea en la obtención de resina alquídicas, en las cuales el ácido
crotónico influye favorablemente en las propiedades de nivelación de las
pinturas preparadas con ellas.
4.4.4.
Acetato de etilo
De los
posibles caminos de síntesis del acetato de etilo, sólo se han empleado hasta
ahora dos procedimientos industriales. La materia de partida, según el país, es
el etanol o el acetaldehído.
En EE.UU.,
donde el alcohol es muy barato, se estatifica con ácido acético en presencia de
un catalizador ácido:
![]()
Si la
transformación tiene lugar en una columna de funcionamiento continuo, se
obtiene un rendimiento del 99%. El acetato de etilo también se forma, junto a
otros numerosos productos, en la oxidación de n-butano (ver sección 4.4.1.2) y
se le puede aislar económicamente, como,
por ejemplo, por la UCC en EE.UU.
En otros
países, en los que se dispone de suficiente acetaldehído como en Japón o en
Alemania Federal, o donde el etanol se encarece por impuestos fiscales, se
emplea preferentemente, como proceso de obtención, la reacción de Tischenko con
acetaldehído:
![]()
Como
catalizador se emplea una disolución de alcoholato de aluminio en una mezcla de
etanol/acetato de etilo con iones de Zn+ y CI- como
promotores. En esta disolución y con enfriamiento a 0-5ºC se produce la
reacción exotérmica del acetaldehído a acetato de etilo. Para una
transformación del 95% se alcanza una selectividad del 96% (etanal). Como
subproducto se forma aldol, que fácilmente se deshidrata. El agua así producida
da lugar a la hidrólisis del catalizador, el alcoholato de aluminio, que
produce su desactivación rápidamente. Por tanto, los procesos fundados en la
adición de acético al etileno, que hasta ahora sólo se describen en patentes,
adquirirán mayor importancia en el futuro:
![]()
El acetato de
etilo es un disolvente importante, que se utiliza sobre todo en la fabricación
de pinturas.
4.4.5. Piridina y alcohilpiridinas
Las bases
piridínicas de importancia industrial son la propia piridina, la
2-metilpiridina (2-picolina) y 2-metil-5-etil-piridina (MEP). La 3 y
4-picolinas tienen sólo una aplicación limitada. La piridina actualmente aún se
obtiene principalmente del alquitrán de hulla, en el que junto con derivados
más volátiles se encuentra en un 0,1% en peso. Sin embargo, las síntesis de las
alcohilpiridinas a causa de su creciente demanda van ganando importancia.
La capacidad
mundial de piridinas de síntesis se estimó en 1972 en 36 000 toneladas anuales.
En Europa, el único productor de piridinas sintéticas (en 1977) es la Reilly Chemicals de Bélgica.
De los
numerosos procesos conocidos, los que han conseguido la mayor importancia son a
base de acetaldehído sólo o junto con formaldehído y amoniaco.
La obtención
de 2-metil-5-etilpiridina es la más sencilla y al mismo tiempo la reacción que
transcurre con una selectividad suficientemente económica, por lo que también
ha sido la primera que ha encontrado aplicación industrial. En el proceso
continuo se hace reaccionar el acetaldehído (directamente o en forma de
paraldehído) con una disolución acuosa de amoniaco al 30-40%. La conversión
tiene lugar en la fase líquida a 220-280ºC y 100-200 bars en presencia de
acetato amóniaco como catalizador:

El producto de
reacción forma dos fases. La fase acuosa
es, en su gran parte, reciclada para
nueva reacción y la fase
orgánica se elabora por medio de destilaciones de azeótropos y en vacío.
La
selectividad en 2-metil-5-etilpiridina alcanza el 70% (acetaldehído). Como subproducto se obtienen 2 y 4-picolinas en
una proporción de 3:1 y bases piridínicas superiores
Si se realiza
en fase gaseosa con AI2O3 o AI2O3 .
SiO2 como catalizadores, que además pueden contener otros
promotores, se forman preferentemente 2 y 4-picolinas en idéntica proporción a
partir de acetaldehído y amoniaco a presión
normal y 350-500ºC.
Si se emplea
una mezcla de acetaldehído y formaldehído junto con NH3 en fase
gaseosa, se obtiene piridina y 3-picolina:

La proporción
en que se emplean los aldehídos determina la proporción de los productos de
reacción. También esta vía ha encontrado aplicación industrial.
La DMS ha
puesto en 1977 en funcionamiento la producción de 2-picolina en una nueva
instalación por selectivo y en varias, etapas con capacidad anual de 3000
toneladas. En el primer paso se hace reaccionar catalíticamente en medio básico
(isopropilamina) la acetona con el acrilonitrilo, con lo que se produce el
nitrilo del ácido 5-oxo-hexanoico con
selectividad superior al 80% (acetona, acrilonitrilo). Éste, finalmente, por
hidrogenación, en presencia de catalizadores metálicos depositados en soportes,
por ejemplo, Ni/SiO2 o Pd/AI2O3, en fase
gaseosa se cicla a 2-picolina o a sus
derivados hidrogenados con eliminación de agua:

Un nuevo
proceso de obtención de 2-picolina y 2-metil-5-etilpiridina de la Nippon Steel
Chemical Co. Hasta ahora no ha tenido
aplicación industrial, pero es diferente a todos los procesos conocidos, ya que
parte de etileno. Éste, junto con amoniaco en presencia de disolución amoniacal
de sales de paladio y con un sistema redox de Cu a 100-300ºC y 30-100 bars,
produce ambas alcohilpiridinas con una selectividad en conjunto del 80%
(etileno):

La principal
aplicación de la 2-metil-5-etilpiridina es en la obtención del ácido nicotínico
(piridín-3-carboxílico, Niacin) por oxidación con ácido nítrico y
descarboxilación selectiva del grupo carboxilo en posición 2:

El ácido
nicotínico y sus derivados, por ejemplo la nicotinamida, forman parte del
complejo vitamínico B. Tienen preferentemente importancia farmacéutica y
son aditivos para alimentos y piensos.
La nicotinamida se obtienen por reacción de los ésteres nicotínicos con NH3.
Otra posible vía es la tantas veces investigada amonoxidación de la 3-picolina
a nitrilo nicotínico y posterior hidrólisis parcial a nicotinamida:

Excepto en una
planta piloto de Degussa, hasta ahora no ha encontrado aplicación industrial.
La demanda mundial de ácido nicotínico y de sus derivados es, por ahora, de
unas 10 000 toneladas al año, con un 35% en EE.UU., el 25% en Europa occidental
y el 10% en el Japón.
La 2-picolina
es un producto de partida para la obtención de 2-vinilpiridina, un comonómero,
que junto con el butadieno y el estireno da copolímeros para la cohesión entre
las fibras de síntesis y el caucho en la industria de cubiertas para
automóviles. Los derivados de la piridina encuentran también aplicación en la
síntesis de herbicidas y en numerosos productos farmacéuticos.