Síntesis con
monóxido de carbono
(Ir a página principal http://organica1.org/ )
Síntesis con monóxido de carbono. 1
3.1. Hidroformilación de
olefinas
3.1.1. Fundamentos
químicos de la hidroformilación.
3.1.2. Sistema
industrial de hidroformilación.
3.1.3. Variantes en la
catálisis de la hidroformilación
3.1.4. Aplicación de los
productos oxo
3.1.4.2 Ácidos
carboxílicos oxo
3.1.4.3. Productos de
aldolización y condensación de los aldehídos oxo
3.2. Carbonización de
olefinas
3.3. Síntesis de ácidos
carboxílicos según Koch
El monoxído de carbono se ha descrito ya en el capítulo <<Productos básicos de la síntesis industrial>>, por su importancia económica, como
componente para síntesis en forma pura o en mezcla con hidrógeno, por ejemplo,
para la obtención de metanol que es ponderalmente su más importante derivado.
Otras transformaciones en que se emplea CO en procesos industriales, como, por
ejemplo, con acetileno para el acrílico (ver apartado 11.1.7.1), con metanol
para acético (ver apartado 7.4.1.3), con propeno para butanol (ver apartado 8.1.3),
son tan específicos, que se describirán especificamente más adelante. En este
capítulo se tratarán reacciones de CO como la hidroformilación, carbonilación y
reacción de Koch, que constituyen un amplio intervalo de transformaciones de
las olefinas.
3.1.
Hidroformilación de olefinas
La hidroformilación o síntesis-oxo es un proceso empleado en la gran
industria para la obtención de aldehídos a partir de olefinas, monóxido de
carbono e hidrógeno.
El fundamento de la reacción fue descubierto en 1938 por O. Roelen en la
Ruhrchemie, cuando observó, al hacer reaccionar etileno con CO y H2 a
presiones y temperaturas elevadas, con catalizadores que contenían cobalto y
torio, que se formaba aldehído propiónico. La hidroformilación se desarrolló
con relativa rapidez en los años siguientes, hasta llegar a ser un proceso
industrial para la fabricación de alcoholes detergentes con longitudes de
cadena de C12 hasta C14, de forma que ya en 1945 entró en
funcionamiento la primera instalación de síntesis-oxo con una producción anual
de 10 000 toneladas.
Mientras tanto, la hidroformilación ha alcanzado en todo el mundo una
sorprendente importancia. La capacidad mundial de todos los productos de
hidroformilación llegó a mitad de 1977 a los 4,1 millones de toneladas al año, con
un pronóstico de aumento para los próximos años hasta los 6,5 millones de
toneladas. La olefina más importante empleada como producto de partida es el
propeno, que da como productos finales más significativos el n-butanol y 2-etilhexanol. El foco de las
actividades de hidroformilación está en Europa occidental, con el 53% de la
capacidad mundial (30% América del Norte, 10% Asia, 5% Europa oriental, 1%
Australia, 1% América latina).
La Alemania Federal aportó, en 1977, 1,1 millones de toneladas de productos
oxo. Los productores son la BASF, RCH (Ruhrchemie) y Hüls.
3.1.1.
Fundamentos químicos de la hidroformilación
La hidroformilación se puede realizar con un gran número de olefinas de
cadena lineal o ramificada con enlaces dobles terminales o internos.
Prácticamente tienen importancia, como productos de partida, las olefinas con 2
hasta con 20 átomos de carbono. La hidroformilación produce, de forma
característica, siempre mezclas de aldehídos isómeros excepto el etileno que da
en principio sólo propionaldehído- a causa del enlace del grupo carbonilo a uno
u otro de los átomos de carbono del enlace doble:
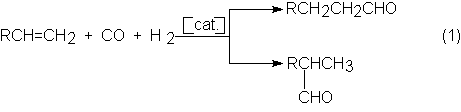
En la mezcla de isómeros el n-aldehído
predomina sobre el i-aldehído.
Además, el curso de la
hidroformilación está influido por la estructura y magnitud molecular de la
olefina empleada. Aun cuando el oxo-aldehído obtenido, puede, en cambio, el
doble enlace interno emigrara una posición terminal, por lo que pueden aparecer
otros componentes en la mezcla de aldehídos. Si se procede con suficiente temperatura
y tiempo de permanencia, el equilibrio de los isómeros sobre el catalizador de
hidroformilación se establecerá por completo (posición terminal del doble
enlace), por lo cual los isómeros de doble enlace de esta olefina pueden
proporcionar el mismo oxo-aldehído.
Además, las ramificaciones en las olefinas, en general, disminuyen la
velocidad de reacción y especialmente cuando la ramificación se encuentra en el
átomo de carbono del doble enlace.
La ramificación a ambos lados del doble enlace puede dificultar fuertemente
la hidroformilación como en el caso del 2,3-dimetil-2-penteno.
La hidroformilación es fundamentalmente una reacción homogénea catalizada y
como tal transcurre a elevadas presiones y temperaturas (en su mayoría 200
hasta 450 bars y 100-200ºC).
Como catalizadores se utilizan combinaciones o complejos de Co, Rh o Ru,
cuya actividad y selectividad se pueden
modificar con ligandos aminados o fosfinados. Aunque en los libros se citan
promotores adicionales, en la práctica no han alcanzado ninguna importancia.
En los procesos industriales se emplean preferentemente compuestos de Co
que comparativamente son más baratos y tienen gran actividad. En las
condiciones de reacción se obtiene entonces como forma activa del catalizador
el hidruro de cobaltotetracarbonilo HCo(CO)4 que se encuentra en
equilibrio con el dicobaltooctocarbonilo Co2(CO)8.
En el transcurso de la reacción, el hidruro de cobaltotetracarbonilo se
transforma en hidruro de cobaltotricarbonilo, con un espacio de coordinación
libre que ocupa después la olefina por formación de un complejo-p.
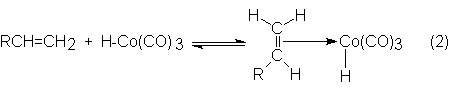
Éste se ordena por formación de un enlace C-Co en un complejo-s, el cual se satura con CO para dar un alcohilcobaltotetracarbonilo:
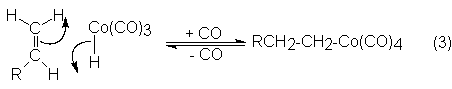
En el paso próximo se reordena el complejo alcohilcobalto tetracarbonilo a
complejo acilcobaltotricarbonilo, el cual por hidrogenación forma un aldehído
por disociación y se produce de nuevo el hidruro de cobaltotricarbonilo:
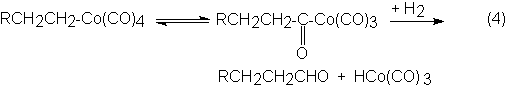
En el transcurso de la reacción no se pueden separar entre sí las
reacciones parciales mencionadas. Se producen simultáneamente y además se
superponen las formaciones de isómeros, de forma que el enlace cobalto-carbono
que se forma según la ecuación (3), no sólo tienen lugar en el carbono final
del doble enlace, sino también en el
interior.
En la mezcla de isómeros predomina, en efecto, el n-aldehído, pero a causa de su mayor importancia industrial,
comparado con el i-aldehído, todos
los procesos industriales están orientados por medio de modificaciones del
catalizador y por condiciones adecuadas del proceso, a aumentar más aún su
proporción.
Así pues, se puede
influir sobre la proporción n/i dentro de determinados límites por
variación de temperatura y presión parcial de Co (ver apartado 3.1.2), y
decisivamente por modificación del catalizador.
En las catálisis con complejos metálicos se puede modificar el catalizador
por cambio de los ligandos y del átomo central. Ambas posibilidades se emplean
en la práctica y han llevado a los siguientes resultados importantes:
·
Los complejos que forman aditivos, como las aminas terciarias, los fosfitos
o, especialmente, las fosfinas, como, por ejemplo, tributil o trifenilfosfina,
aumentan la proporción de contenido n con disminución de la velocidad de
hidroformilación y menor selectividad en aldehído.
·
El empleo de rodio, en lugar de cobalto, como catalizador produce una
disminución de la porción n, pero, en
cambio, elevada la velocidad de hidroformilación y la selectividad de aldehídos
n/i
(ver apartado 3.1.3).
La influencia del ligando en la proporción n/i se explica por la
disminución de la densidad electrónica en el átomo central, como también por el
volumen espacial que ocupa la molécula del catalizador; los ligandos de
fosfinas muy voluminosos dificultan el ataque a los átomos de C interiores, es
decir, elevan la proporción n en el
producto de reacción.
Con una adecuada combinación de rodio como átomo central y con fosfinas
como ligandos, se pueden mejorar los efectos positivos, de tal forma que se
puede alcanzar una alta proporción n/i. Así se obtiene, partiendo de propeno,
la proporción n- e i-butiraldehído que varía de 8-16:1
(prácticamente es frecuente obtener una proporción de alrededor 10:1) frentea
8:2 con un catalizador de cobalto no modificado. Otras ventajas, como la menor
presión de reacción y sencillez en la elaboración del producto de reacción,
están contrapesadas por la menor actividad del catalizador y el elevado precio
del rodio.
Dejando aparte la formación de i-aldehídos
como productos secundarios no deseados y la mencionada isomerización de los
enlaces dobles de las olefinas, que son consustanciales con el principio de la
hidroformilación, hay además otras reacciones secundarias y subsiguientes que
modifican la selectividad en n-aldehídos,
según el tipo de catalizador y condiciones de reacción.
Entre las reacciones secundarias se encuentra la hidrogenación de las
olefinas empleadas a hidrocarburos saturados. A las reacciones subsiguientes
corresponde la rehidrogenación de los n e i-aldehídos
a los correspondientes alcoholes, así como condensados aldólicas, formación de
ésteres fórmicos por <<hidroformilación dealdehído>> y acetalización, por lo que hay un
gran número de problemas a resolver si se quiere alcanzar la máxima
selectividad en aldehídos lineales.
3.1.2.
Sistema industrial de hidroformilación
Las formas industriales de realizar
la hidroformilación de olefinas se basan actualmente con combinaciones de cobalto, como catalizadores
sin adición de fosfinas o fosfitos (ver
<<Variantes en la catálisis>>, apartado 3.1.3).
Veamos, pues, como se desarrollan las distintas etapas del proceso
industrial, con cobalto como catalizador, en el caso, como ejemplo, de la
hidroformilación del propeno, que es la olefina de partida de mayor producción.
Hay que distinguir las tres etapas siguientes:
·
Hidroformilación (incluida la preparación de catalizador).
·
Separación del catalizador (incluida su elaboración).
·
Aislamiento de los productos de reacción.
Para la 1. Etapa del proceso:
El cobalto se introduce en el reactor de alta presión de acero
inoxidable, como polvo metálico, hidróxido o sal. Es aquí donde reacciona en
fase líquida, formada por el propeno y productos de la oxorreación, en las
condiciones de hidroformilación a 250-300 bars y 140-180ºC con el oxo-gas (H2
+ CO), produciéndose rápidamente el hidruro de cobaltocarbonilo. Generalmente
el producto de reacción de la olefina sirve como disolvente, pero también se
puede emplear una mezcla de alcalinos.
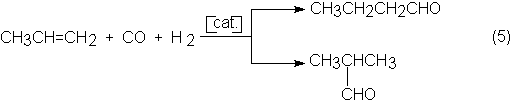
El propeno reacciona entonces con H2 y CO para
dar una mezcla de n e i-butiraldehído:
El calor de
reacción, de unas 28-35 kcal (118-147)/mol de olefina, se elimina por un
enfriador tubular.
El producto bruto condensable está compuesto
aproximadamente por un 80% en peso de butiraldehídos, 10-14% de butanoles y
formiatos de butilo, 6-10% de otras sustancias, como, por ejemplo, las de
ebullición más alta. La relación n a i-butiraldehído es de aproximadamente
75:25 a 80:20. La mezcla CO/H2 se transforma hasta alrededor del 90%
en aldehídos y alcoholes aislables; el resto se elimina con inertes y se quema.
La selectividad
en productos C4 alcanza el 82-85% (C3H6).
El 15-17% del propeno convertido se encuentra en los productos de ebullición
alta o bien como propano en el gas residual.
La transformación y selectividad dependen, en forma compleja,
de numerosas variables del proceso. Apoyándonos en una ecuación de velocidades
propuesta por G. Natta vamos a exponer simplificadamente algunas influencias de
la concentración. La deseable alta velocidad de formación de un aldehído para
su obtención industrial depende de los valores altos de las concentraciones y,
respectivamente, del cociente de las presiones parciales:
![]()
Esto se puede alcanzar en principio de dos formas:
·
Por una presión parcial de CO pequeña.
·
Por concentraciones altas de olefina y cobalto, así como también por una
presión parcial alta de H2.
Paso 1:
Una presión parcial baja de CO acelera, en efecto, la
hidroformilación; se demuestra, sin embargo, que es necesario mantener una
presión mínima de CO, dependiente de la temperatura de reacción, para mantener
estable el catalizador HCo(CO)4 y con ello su actividad.
Paso 2:
Si, por ejemplo, se eleva la presión parcial de H2
en el gas-oxo o la concentración de catalizador, aumenta, en efecto, la
velocidad de formación, es decir, la transformación del propeno en la forma
deseada. Sin embargo, se intensifica la hidrogenación a alcohol y la formación
de propano, lo que hace disminuir simultáneamente la selectividad de formación
de n-butiraldehído. Por ello, para
alcanzar una selectividad alta, se debe disminuir la transformación de propeno.
Este modo de actuar llega de todas formas pronto a su límite al no ser
económico el rendimiento por volumen y tiempo. Otras variables del proceso, con
interacciones recíprocas, complican el llegar a mejorarlo.
Un problema todavía no satisfactoriamente resuelto de la
hidroformilación del propeno es la forzosa formación de i-butiraldehído, que no siempre encuentra un empleo económico
adecuado. Se han desarrollado modificaciones del proceso de hidroformilación,
sobre todo, con otros sistemas de catalizadores que principalmente producen una
elevación de la selectividad en n-butiraldehído.
Para la 2. Etapa del proceso:
Para la
separación del hidruro de cobaltocarbonilo de los productos líquidos de
reacción se han desarrollado principalmente dos procesos, con modificaciones
especiales usadas por cada fabricante de productos oxo. Por uno de estos
métodos se calienta la mezcla de reacción, después de reducir la presión a unos
20 bars. Se precipita así un barro de cobalto, que se separa, y tras proceder a
regenerarlo se lleva de nuevo al reactor (como en el proceso de la Ruhrchemie).
Otra separación del cobalto se aplica sobre todo cuando
se producen aldehídos inferiores; el cobalto se recupera, bien por un tratamiento
ácido (con acético en el proceso de la BSF, ácidos carboxílicos superiores en
el proceso Mitsubishi, H2SO4/CH3COOH en el
proceso UCC) en presencia de aire o de O2 en forma de disolución
acuosa de sales de Co o como Co(OH)2 por precipitación con hidróxidos
alcalinos, o también como hidrocarbonilo por extracción con una disolución de
bicarbonato sódico (como en el proceso Kuhlman). Después de acidularlo, se
extrae de nuevo con la olefina empleada o con sustancias auxiliares y se
recicla en el reactor para continuar el proceso.
Para la 3. Etapa del proceso:
El producto de reacción de Co se separa por destilación a
la presión normal.
Primeramente, tiene lugar el aislamiento de una
mezcla de n e i-butiraldehído que,
finalmente -a causa de la pequeña diferencia
en los puntos de ebullición de 10ºC-, se tiene que fraccionar en sus
componentes puros por medio de una columna de rectificación muy eficaz. El
residuo de la separación de los aldehídos contiene n- e i-butanoles formados
en la oxo-reacción por hidrogenación de los aldehídos, así como otros productos
secundarios, como formiatos, acetales y los llamados aceites pesados. La mezcla
residual, directamente o con tratamiento previo, por ejemplo, hidrólisis, se
hidrogena a butanoles. Si se hidroformilan olefinas superiores al propeno, se
renuncia generalmente a separar los aldehídos y se hidrogena el producto bruto
directamente después de separar el catalizador de cobalto, con lo que se
obtiene la mezcla de n- e i-alcoholes.
3.1.3.
Variantes en la catálisis de la hidroformilación
Para disminuir la formación de i-butiraldehído
ha empleado la Shell, en plantas oxo, catalizadores del tipo HCo(CO)3.
P(n-C4H9)3
a presiones bajas de 50-100 bars y 180-200ºC.
El catalizador, sin embargo, por su contenido de fosfina relativamente
alto, tiene una menor actividad y selectividad que el hidruro de
carbonilcobalto sin modificar. A pesar de la mejor relación n/i
( un 90% de producto lineal), la selectividad en aldehído total disminuye,
puesto que las reacciones secundarias y subsiguientes, como la hidrogenación de
la olefina empleada a hidrocarburo saturado, así como, sobre todo, la de los
aldehídos a sus correspondientes alcoholes aumentan considerablemente.
Por ello, el proceso de la Shell es apropiado cuando los alcoholes son los
productos oxo preferidos.
Otra variante de catálisis de la hidroformilación consiste en el empleo de rodiocarbonilos solos o
junto con fosfinas, en lugar de los catalizadores de cobalto convencionales.
El mecanismo de la hidroformilación catalizada por hidrocarbonilo de rodio
y trifenilfosfina se formula muy semejantemente a la de la catálisis con
cobalto; se supone como combinación de iniciación, importante para el
transcurso de la acción catalítica, un complejo de hidrocarbonilo de rodio y fosfina,
en el cual, por intercambio de un ligando trifenilfosfina por una molécula de
olefina, se forma finalmente una combinación de n-alcohilrodio:
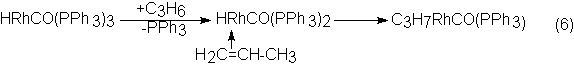
la interposición de monóxido de carbono en el enlace C-Rh e hidrogenólisis
final, completan el ciclo de la catálisis:
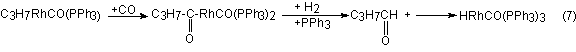
Aun cuando numerosas empresas desde hace muchos años se han ocupado del
desarrollo semiindustrial de la hidroformilacióncatalizada con rodio, sólo se
consiguió la producción industrial en el año 1975.
La UCC junto con la Johnson Matthey y Davy Powergas emprendieron en 1975 la
construcción de una instalación, con una capacidad anual de 70 000 toneladas de
aldehído propiónico (1977) y en 1976 otra instalación con capacidad anual de 136 000 toneladas de n-butiraldehído, a base de catalizadores
de trifenilfosfinarodio. Con esto se consigue por primera vez las ventajas de
una alta proporción de selectividad en el dominio 8-16:1 en n/i-butiraldehídos,
con empleo de bajas presiones de reacción, entre 7 y 25 bars a 90-120ºC, y una
elaboración simplificada de los
productos de reacción -a causa de la mayor estabilidad de los rodio-carbonilos
modificados y a la ausencia de alcoholes C4 en la mezcla de
productos que formarían acetales-, y
todo ello industrialmente (ver apartado 3.1.1).
Mientras tanto, también otras empresas han puesto en funcionamiento plantas
industriales basadas en la tecnología del rodio de UCC (Johnson Matthey y Davy
Powergas) o las tienen proyectadas.
3.1.4.
Aplicación de los productos oxo
La hidroformilación proporciona, como productos primarios, aldehídos. Por
ello, se designa este principio de síntesis reacción oxo y casi todos los
aldehídos obtenidos, así como sus derivados se reúnen bajo la denominación de
productos oxo.
Los oxoaldehídos como producto final no tienen prácticamente importancia.
Sin embargo, como combinaciones relativas, son productos intermedios
importantes para la obtención de alcoholes oxo, ácidos carboxílicos oxo,
así como productos de condensación
aldólica. Los oxoaldehídos se transforman en pequeña cantidad en aminas
primarias por aminación hidrogenante:
![]()
3.1.4.1.
Alcoholes oxo
Los alcoholes oxo se obtienen por hidrogenación catalítica de los aldehídos
oxo. En principio, puede emplearse también como catalizador el hidruro de
cobaltocarbonilo pero a temperaturas superiores a las de hidroformilación; sin
embargo, se prefieren, generalmente, catalizadores selectivos de Ni o Cu. En la
hidrogenación se emplean generalmente, los aldehídos destilados, libres de
cobalto, aunque se pueden emplear también los productos brutos de la reacción
oxo libres de cobalto.
La hidrogenación se puede realizar bien en fase gaseosa, con catalizadores
de Ni a 2-3 bars y 115ºC o con catalizadores de Cu a temperatura de 130-160ºC y
presión de 30-50 bars, así como también en la fase líquida a 80 bars y 115ºC con catalizadores de Ni:
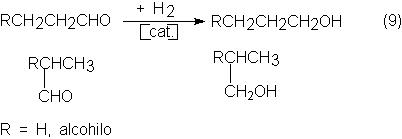
Si las condiciones de hidrogenación se hacen más intensas, por ejemplo,
200ºC y 280 bars, se pueden aplicar
también los productos brutos de la reacción oxo, que, por ejemplo, contienen
formiato de butilo y dibutilacetales
del butiraldehído. Ésteres y acetales, que de otra forma se tendrían que
saponificar por separado, pueden proporcionar cantidades adicionales de
butanoles por hidrogenólisis:

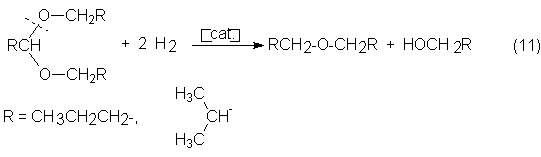
Los alcoholes oxo de longitud de cadena C4-C6 se
pueden utilizar ya directamente o después de esterificación con ácidos
carboxílicos (por ejemplo, acético), sobre todo como disolventes para la industria de pintura y de plásticos.
Los alcoholes oxo C8-C13 que se pueden obtener de
oligómeros de olefinas (por ejemplo, i-heptenos,
di-i-butenos, tripropenos) y de
olefinas de disociación se emplean, preferentemente, esterificados por ácidos
dicarboxílicos o sus anhídrido (anhídrido ftálico), como plastificantes (ver
apartado 14.1.3).
También las olefinas superiores, ramificadas o no, accesibles
industrialmente, proporcionan, por hidroformilación e hidrogenación, alcoholes
C12-C19 importantes, empleados para la fabricación de
tensoactivos y materias auxiliares para la industria textil.
El alcohol oxo inferior, cuantitativamente más importante, es el n-butanol. Las cantidades de producción
de n-butanol en los países
industriales más importantes se dan en la tabla al margen. Para la producción
total de todos los butanoles (n + I), ver apartado 8.1.3.
Alrededor del 50% de los butanoles producidos se utilizan directamente o
esterificados con ácidos carboxílicos, como, por ejemplo, acético, butírico,
valeriánico, glicólico o láctico, como disolventes para grasas, aceites, ceras,
resinas, naturales y plásticos de síntesis. Los ésteres de n-butilo tienen además otras aplicaciones, por ejemplo, el
acrilato de n-butilo junto con otros comonómeros se utiliza para preparar
dispersiones. El ftalato de di-n-butilo
(DBP) fue mucho tiempo un producto normalizado como plastificante para el
cloruro de polivinilo (PVC).
Como en el caso del n-butanol, la
producción de i-butanol se consume en
aprte considerable en el sector de disolventes, puesto que su poder disolvente
es muy semejante al del n-butanol. El
ftalato de di-i-butilo (DIBP) ha
encontrado, como el DBP, aplicación también como plastificante.
3.1.4.2
Ácidos carboxílicos oxo
Los aldehídos oxo se pueden transformar en ácidos carboxílicos mediante agentes oxidantes suaves; en el caso más
simple, con aire. La oxidación se puede llevar a cabo catalíticamente en
presencia de sales metálicas, así como también sin catalizadores a temperaturas
hasta unos 100ºC y presiones hasta 7 bars:
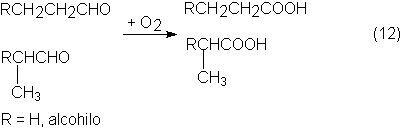
Como metales se emplean
especialmente aquellos que presenten un cambio de valencia, como Cu, Fe, Co,
Mn, entre otros.
Los ácidos carboxílicos así obtenidos se transforman en su mayoría en
ésteres, que con gran amplitud se emplean como disolventes.
Como ejemplos de aplicaciones de ácidos carboxílicos oxo se indican algunos
casos característicos: el n-butírico
para la obtención de acetobutirato de celulosa, un éster mixto que se puede
procesar para recibrimientos resistentes a la humedad, luz y calor. El ácido
isooctanoico y el isononanoico son adecuados para modificar las resinas
alquídicas, y por esterificación con etilenglicoles, como plastificantes para
PVC. Sus sales de Co, Mn, Pb, Zn y Ca se emplean como aceleradores de secado
(secantes) de pinturas, mientras que sus ésteres vinílicos son productos de
partida para obtener dispersiones.
3.1.4.3.
Productos de aldolización y condensación de los aldehídos oxo
Un tercer tipo de reacción secundaria de los aldehídos oxo que se emplean
industrialmente es la aldolización. Para ello, se transforman los aldehídos oxo
en presencia de catalizadores básicos en fase líquida. Del producto primario,
el aldol, por subsiguiente hidrogenación del grupo aldehído, se pueden obtener
los dioles, pero también se puede por deshidratación (crotonización) y
subsiguiente hidrogenación del doble enlace y función aldehído, obtener
alcoholes primarios con doble número de átomos de carbono que el aldehído
empleado. Ambas posibilidades se emplean industrialmente:
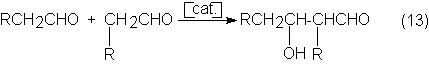
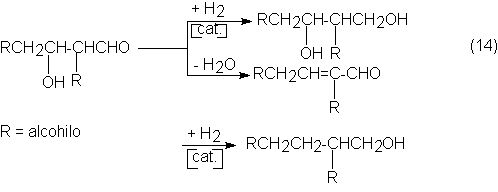
Al grupo de dioles primeramente mencionado pertenece el
2,2,4-trimetilpentano-1,3-diol que se obtiene a partir del i-butiraldehído, y que sólo tiene importancia industrial limitada.
Al grupo segundo de monoalcoholes primarios correspondientes el 2-etilhexanol,
que junto con el n-butanol son los
dos productos oxo que se obtienen en mayor cantidad. En 1976 la capacidad total
mundial era de unos 1,5 millones de toneladas de 2-etilhexanol, de las cuales
a Europa occidental le corresponden
0,69 millones, a Alemania Federal 0,47 millones, a EE.UU. 0,42 millones y al
Japón 0.22 millones. En Alemania los tres productores, BASF, Hüls y RCH,
obtuvieron en 1976 unas 336 000 toneladas de 2-etilhexanol.
Los números de producción en otros años están en la tabla al margen.
La condensación aldólica del n-butiraldehído,
pasando por la crotonización a 2-etilhexenal, con posterior hidrogenación de
éste, da 2-etilhexanol:
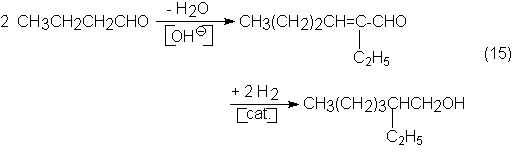
Para ello, el n-butiraldehído se
transforma en 2-etilhexenal con deshidratación
simultánea en presencia de hidróxido sódico o con un cambiador de iones
básicos de forma cuantitativa.
Sigue después la hidrogenación en fase gaseosa con una ligera sobrepresión
hasta de 5 bars y 100-150ºC sobre un catalizadoren lecho estático con níquel o
a 135-170ºC con cobre, con lo que se pasa de 2-etilhexenal a 2-etilhexanol, que
eventualmente se puede seguir hidrogenado en fase líquida si es necesario. El
producto puro se obtiene por una destilación en tres etapas. La selectividad alcanza el 95% (n-butiraldehído).
Otra variante del proceso, que hasta ahora ha sido sólo utilizada por la
Shell en EE.UU. e Inglaterra, y la Exxon en EE.UU. y Japón, consiste en la
combinación de la aldolización y reacción oxo en una sola etapa, que se ha
denominado proceso aldox. Por adición de cocatalizadores como combinaciones de
Zn, Sn, Ti, Al o Cu o también de KOH,
al propio catalizador oxo, se pueden realizar simultáneamente los tres pasos
necesarios hasta la obtención del 2-etilhexanol, es decir, hidroformilación de
propeno, condensación aldólica e hidrogenación.
La obtención de 2-etilhexanol a aprtir de acetaldehído (ver apartado 7.4.3)
actualmente no tiene importancia: una pequeña producción por esa vía existe aún
en los EE.UU. El 2-etilhexanol es, sobre todos los demás alcoholes superiores,
el de mayor importancia económica. Se usa sobre todo para la obtención de
ésteres con ácidos dicarboxílicos, como el ftálico o el adípico.
El producto que se obtiene por reacción de 2-etilhexanol y anhídrido
dtálico, el di-2-etilhexilftalato (DOP) es un excelente plastificante de la
industria de plásticos, fisiológicamente inofensivo. El DOP ha encontrado una
nueva aplicación como líquido dieléctrico para condensadores, con lo que ha
sustituido a los difenilos policlorados, que sí son tóxicos.
Otros ésteres del 2-etilhexanol, especialmente los de ácidosdicarboxílicos
alifáticos, se emplean como aceites hidráulicos o como componentes de
lubricantes sintéticos.
El 2-etilhexanol se puede transformar aún por oxidación en ácido
2-etilhexanóico. El ácido se puede también obtener por hidrogenación selectiva
de 2-etilhexenal, en presencia de catalizadores de Pd, a 2-etilhexanal, cuya
oxidación lo produce. Se usa para modificar las resinas alquídicas. En 1974 se
produjeron en Europa occidental
unas 9200 toneladas de ácido
2-etilhexanóico.
3.2.
Carbonización de olefinas
La carbonilación de olefinas con CO, junto con un reactivo nucleófilo que
contenga átomos de H activos, produce en presencia de carbonilos metálicos
ácidos carboxílicos o sus derivados, como ésteres, tioésteres, amidas,
anhídridos, entre otros:
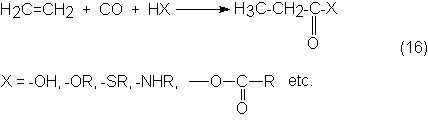
Este tipo de reacción de carbonilación corresponde a las que usan como
catalizadores carbonilos metálicos (por ejemplo, carbonilos de Ni, Co, Fe, Rh,
Ru, Pd) y que constituyen el amplio campo de reacciones de Reppe.
La transformación de las olefinas
con CO y H2O a ácidos carboxílicos se denomina, por semejanza
a la hidroformilación, hidrocarboxilación.
La reacción de Koch, que se describe en el apartado 6.3, es un tipo
semejante de hidrocarboxilación, que parte de los correspondientes componentes,
como olefina, CO y H2O, pero por empleo de un catalizador protónico
y condiciones de reacción más suaves, conduce predominantemente a los ácidos
carboxílicos terciarios.
Un ejemplo de realización industrial de hidrocarboxilación, en condiciones
de Reppe, es la transformación del etileno en ácido propiónico con CO y H2O.
La reacción de los componentes tiene lugar a 200-240 bars y 270-320ºC en fase
líquida, compuesta por ácido propiónico bruto y propionato de Ni disuelto como
catalizador:
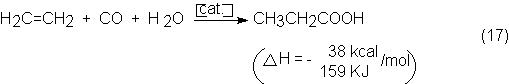
Como catalizador activo resulta el Ni(CO)4 que se forma in situ. También otros carbonilos, por
ejemplo, de Fe o Co son activos catalíticamente. Terminada la reacción, el
Ni(CO)4 contenido en el producto bruto se reconvierte por oxidación
con aire en presencia de propiónico, en propionato, y se vuelve al reactor para
nuevo uso. El rendimientoen ácido propiónico alcanza el 95% (C2H4).
Como productos secundarios se forman CO2, etano y ácidos
carboxílicos superiores, que por su principio de formación tienen un número
impar de átomos de carbono, como, por ejemplo, el valeriánico.
La BASF realiza esta reacción desde 1952 en una instalación industrial que
en 1974 tenía una capacidad de unas 30 000 toneladas al año.
El ácido propiónico se emplea principalmente como conservante en la
industria alimenticia y para la preparación de piensos. Otra aplicación es la
obtención es la obtención de ésteres, como el propionato de amilo, qie sirve
como disolvente para resinas y derivados celulósicos.
Si en la hidrocarboxilación, según Reppe, se emplean olefinas superiores,
se forma , por isomerización simultánea de los enlaces dobles, una mezcla de
ácidos carboxílicos. Así pues, esta vía de obtención queda limitada a la
reacción de etileno.
3.3.
Síntesis de ácidos carboxílicos según Koch
La transformación de olefinas con CO y H2O a ácidos carboxílicos,
además de hacerse por catálisis con carbonilos metálicos (ver Carbonilación de Reppe, apartado 3.2), puede también realizarse con
catalizadores protónicos. Son adecuados, sobre todo, los ácidos minerales, como
H2SO4, HF, H3PO4 solos o combinados
con BF3 o SbF5, como, por, ejemplo, HF . SbF5.
En el transcurso de la reacción se
adiciona primeramente un protón a la
olefina y se forma un carbocatión, que se estabiliza por isomerización del
doble enlace y reordenamiento del esqueleto carbonado:
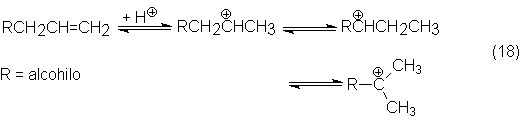
Después se adiciona el CO al carbocatión resultante y se forma de un catión
acilio, que finalmente reacciona con agua para dar ácido carboxílico o con
alcoholes para dar directamente un éster:

Así se obtienen mezclas isómeras de ácidos carboxílicos de cadena
ramificada en las que la porción de ácidos carboxílicos terciarios depende de
las condiciones de la reacción. Así por
ejmplo, todos los isómeros de buteno a unos 80ºC, 20-100 bars y largo tiempo de
reacción, se transforman predominantemente en ácidopiválico (trimetilacético).
En la práctica para obtener este ácido se emplea principalmente i-buteno:
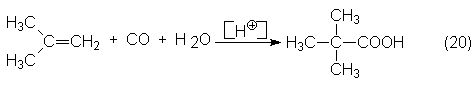
El carbocatión intermedio que se forma puede lograrse con otros productos
de partida; así el i-butanol o el
2-clorobutano pueden emplearse para la obtención de ácido piválico, puesto que
en las condiciones de reacción se desdobla el H2O o el HCI,
respectivamente.
El proceso industrial se realiza en una
sucesión de reactores con agitación a 20-80ºC y 20-100 bars, con CO que se
adiciona a la olefina en presencia de catalizadores, y en la segunda etapa se
añade agua. Como catalizador que proporciona protones se prefiere H3PO4/BF3,
puesto que al añadir el agua se produce la separación de producto y fase con el
catalizador. El H3PO4/BF3
se recicla en el proceso.
Los ácidos carboxílicos terciarios se obtienen con una elevada conversión
de olefinas superiores y con selectividades del 80-100%. Como productos
secundarios resultan los ácidos carboxílicos de las olefinas dimerizadas.Los
procesos industriales fundados en el principio de la reacción de Koch, los
realizan la Shell, Exxon y DuPont. Además del ácido piválico obtenido de i-buteno se obtienen ácidos carboxílicos
de cadena ramificada C6-C11 de las olefinas
correspondientes. Estos ácidos se encuentran en el comercio bajo la
denominación de <<ácidos versáticos>> (Shell) o <<neo-ácidos>> (Exxon).
El comportamiento químico de los ácidos carboxílicos terciarios está determinado por las
ramificaciones alcohólicas en posición a respecto al grupo carboxilo. Esto
conduce a un fuerte impedimento estéreo, que determina la extraordinaria
estabilidad térmica y difícil saponificación de los ésteres, por ejemplo. Son
por ello adecuados como componentes para aceites sintéticos. Los ácidos sirven
para elaborar resinas y lacas. Los éteres vinílicos se emplean como comonómeros
para la obtención de dispersiones o también en copolímeros con VC para
plastificación interna.
Los ésteres glicidílicos de los ácidos de Koch sirven para modificar las
resinas alquídicas.
Los sales metálicas de los ácidos carboxílicos terciarios muy ramificados
se emplean como aceleradores de secado.