Página principal http://organica1.org
25 Reacción de alcoholes
con halogenuros de hidrogeno.
Catálisis ácida
Un método para obtener halogenuros de alquilo se da empleando la
reacción de alcoholes con halogenuros de hidrógeno (haluros7).
Estudiemos con más detenimiento esta reacción, no sólo como método de síntesis
importante, sino como ejemplo de una sustitución nucleofílica.
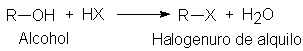
Al hacer esto, veremos algo que resulta completamente nuevo: la manera
de transformar instantáneamente, un grupo
saliente pobre en otro muy bueno, y sin más esfuerzo que el de verte una
solución de una botella en un matraz. Veremos el tipo más importante y más
simple de efecto catalítico que conoce el químico orgánico: uno que tiene un
papel clave en la química de compuestos de todo tipo, tanto en el tubo de ensayos, como en el organismo vivo.
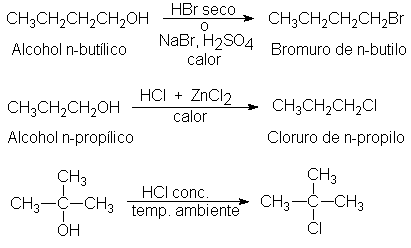
Los alcoholes reaccionan con facilidad con los halogenuros de
hidrógeno para dar halogenuros de alquilo y agua. La reacción se produce
haciendo pasar halogenuro seco por el alcohol o calentado el alcohol con el ácido acuoso concentrado. En presencia
del alcohol, a veces se genera bromuro de hidrógeno mediante la reacción entre
ácido sulfúrico y bromuro de sodio.
El menos reactivo de los halogenuros de hidrógeno, el HCI, requiere,
por lo general, la presencia de cloruro de cinc para reaccionar con alcoholes
primarios y secundarios; por otra parte, el alcohol t-butílico, muy reactivo,
se convierte en el cloruro por simple agitación con ácido clorhídrico
concentrado a temperatura ambiente. Ejemplos:
Enumeremos algunos hechos conocidos acerca de la reacción entre alcoholes y halogenuros de hidrógeno.
(a)
La reacción es catalizada por ácidos. A
pesar de que los propios halogenuros de hidrógeno acuoso son ácidos fuertes, la
presencia de ácido adicional acelera la formación de halogenuros.
(b)
Hay transposición del grupo alquilo, salvo en la mayoría de los
alcoholes primarios.
El grupo alquilo del
halogenuro no tiene siempre la misma estructura que en el alcohol original. Por
ejemplo:
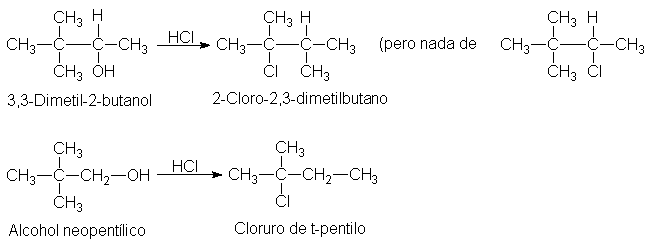
Podemos apreciar que el halógeno no siempre se une al carbono que
originalmente tenía el hidroxilo (primer ejemplo); aún puede ser diferente el
esqueleto carbonato del producto al del material de partida (segundo ejemplo).
Por otra parte, y tal como se ilustró para los alcoholes n-propílico y
n-butílico, la mayoría de los alcoholes primarios dan altos rendimientos en
halogenuros primarios sin
transposición.
(c)
El orden de reactividad de los alcoholes con HX es 3º > 2º > 1º
< CH3. La reactividad decrece a
lo largo de la mayor parte de la serie (este orden es la base del ensayo de Lucas), pasa por un mínimo en 1º y aumenta de nuevo en CH3.
¿Qué nos sugieren los hechos recién enumerados en cuanto al mecanismo
de la reacción entre alcoholes y halogenuros de hidrógeno?
La catálisis por ácido
sugiere que está involucrado el alcohol protonado ROH2+. Las
transposiciones establecen que los carbonationes son intermedios aunque no con
alcoholes primarios. La idea de los carbocationes se apoya en el orden de reactividad de los alcoholes,
que corre paralelo a la estabilidad de los carbocationes, excepto para el
metilo.
Basándonos en estas evidencias, formulamos el mecanismo siguiente:
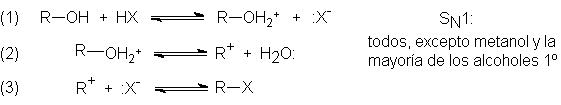
El alcohol acepta el ión hidrógeno para formar la especie protonada
(paso 1), que se disocia en agua y un carbocatión (paso 2); el carbocatión se
combina entonces con un ión halogenuro (no necesariamente con el del paso 1)
para formar el halogenuro de alquilo (paso 3).
Observando el mecanismo expuesto, reconocemos la naturaleza de la
reacción: sustitución nucleofílica,
con el alcohol protonado como sustrato y el ión halogenuro como nucleófilo. Una
vez reconocido el tipo de reacción, las evidencias de la información encajan.
El conjunto de ecuaciones expuesto arriba es, por supuesto, el
mecanismo para la sustitución SN1. Los alcoholes primarios no se
transponen, simplemente porque no reaccionan por este mecanismo, sino por el SN2:
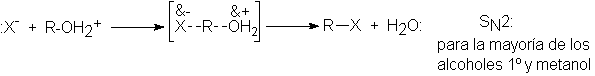
Tenemos aquí otro ejemplo de la característica de la sustitución
nucleofílica: un cambio en la molecularidad de la reacción, en este caso
particular entre 2º y 1º. Se confirma este cambio porque la reactividad pasa
por un mínimo en 1º y aumenta nuevamente en el metilo.
Comenzando con el extremo metilo de la serie, revisemos lo que
probablemente suceda aquí. El sustrato metílico es el menos capaz de heterólisis
y el más abierto para el ataque nucleofílico: reacciona por un mecanismo SN2
total. También lo hacen los sustratos primarios, pero, debido a un mayor
impedimento estérico, reaccionan más lentamente que el metilo. Los sustratos
secundarios presentan un impedimento estérico aún mayor, pero tienen mayor
capacidad para generar carbocationes. La heterólisis es más rápida para ellos
que un ataque nucleofílico por un ión halogenuro, por lo que el mecanismo
cambia aquí a SN1. Con el cambio de
mecanismo comienza a aumentar la velocidad. También los sustratos
terciarios reaccionan por un mecanismo SN1; reaccionan con mayor
velocidad que los sustratos secundarios, debido a la mayor dispersión de carga
en los carbocationes incipientes.
Hasta ahora hemos tratado esta reacción en función de la
clasificación, muy útil, de los sustratos en primarios, secundarios o
terciarios. No obstante, debemos tener presente que esta clasificación como tal
no es lo importante, sino los factores que realmente operan; en esta reacción,
el impedimento estérico al ataque nucleofílico y la dispersión de carga en el carbocation incipiente. Estos factores
dan origen entre otras cosas a la
relación entre 1º, 2º, 3º, y a la
competencia SN2 SN1. Pero hacen más que esto, pueden
hacer que un sustrato de una clase actúe como uno de otra; sin embargo, tal
comportamiento es comprensible con sólo examinar las estructuras implicadas.
Veamos dos de tales ejemplos.
Como se indicó anteriormente, el alcohol neopentílico reacciona con
transposición casi completa, lo que demuestra, a pesar de ser primario, que
sigue un mecanismo carbocatiónico. Esto es contrario a nuestra generalización,
pero fácilmente explicable. Aunque el neopentilo es un grupo primario, es muy
voluminoso, de modo que los sustratos
neopentílicos sufren reacicones SN2 con mucha lentitud (Sec.
5.15). La formación del catión neopentilo es también muy lenta; sin embargo, es
mucho más rápida que la reacción bimolecular alternativa.
Nuestro segundo ejemplo involucra al 1-cloro-2-propanol. Aunque
técnicamente es un alcohol secundario, reacciona con los halogenuros de
hidrógeno de forma anormalmente lenta,
con una velocidad similar a la de un alcohol primario. Esta vez no estamos
tratando con un efecto estérico, sino con uno polar. La velocidad de una
reacción SN1 depende de la
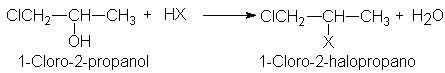
Estabilidad del carbocation que está formando (haluros22).
Comparemos el catión 1-cloro-2-propilo con uno secundario simple, por ejemplo,
el catión isopropilo. El cloro electronegativo ejerce un efecto inductivo de
atracción electrónica. Como sabemos ya (haluros21),
esto intensifica la carga positiva sobre el carbono deficiente en electrones,
por lo que
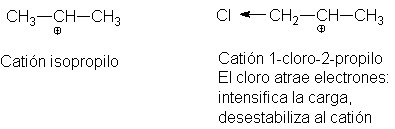
El carbocatión es menos estable. Esta misma atracción de electrones
desestabiliza al catión incipiente en el estado de transición, eleva la Eact
y retarda la reacción.
Estudiemos ahora lo que para nosotros es el aspecto más importante de
la reacción entre un alcohol y los halogenuros de hidrógeno:
la catálisis ácida. ¿Qué hace el ácido? En un primer paso, convierte al
alcohol en la especie protonada, que de hecho es el sustrato sometido a sustitución. En ausencia de ácido,
la sustitución debería implicar por cualquiera de los dos mecanismos la pérdida
de un ión hidróxido, que es fuertemente básico y un saliente muy pobre. La
sustitución con el alcohol protonado como sustrato implica, por otra parte, la
pérdida de agua que es sólo débilmente básica y un grupo saliente muy bueno. La
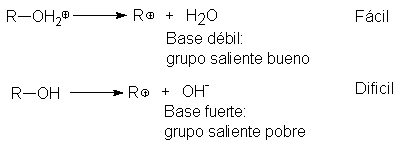
Protonación del alcohol es un simple equilibrio ácido-base, y tiene
lugar instantáneamente al mezclar los reactivos; sin embargo, transforma un grupo
saliente muy malo en uno excelente, permitiendo así que la reacción suceda. Las
pruebas indican que casi nunca ocurre la separación de un ión hidróxido de un
alcohol; las reacciones que involucran la ruptura del enlace C-O de un alcohol
en casi todos los casos parecen requerir un catalizador ácido, cuyo propósito,
como aquí, es la formación de la especie protonada.
Así, los alcoholes sufren sustitución nucleofílica, igual que los
halogenuros de alquilo, tanto por el mecanismo SN2 como por el SN1,
pero los primeros tienden más al unimolecular.
De una forma general, se puede apreciar por qué sucede esto. Para
sufrir sustitución, un alcohol debe estar protonado, lo que necesita un medio
ácido. Hemos visto que una reacción SN2 se ve favorecida por el uso
de un nucleófilo potente, algo que es muy factible en reacciones con
halogenuros de alquilo, pero no podemos tener un nucleófilo fuerte una base fuerte en el medio ácido necesario
para la protonación de un alcohol: cualquier base mucho más fuerte que el propio
alcohol sería protonada a expensas de éste. Por tanto, confinado a reaccionar
con reactivos débilmente básicos y débilmente nucleofílicos, los alcoholes
reaccionan principalmente por medio del mecanismo carbocatiónico.
En el párrafo inicial de esta sección se afirmó que la protonación era
el efecto catalítico más importante y el más simple de la química orgánica. En
presencia de ácidos, muchos tipos de átomos que se encuentran en compuestos
orgánicos se protonan significativamente: oxígeno, nitrógeno, azufre e incluso
carbono. Como veremos en casi todos los capítulos de este hecho, esta
protonación tiene efectos poderosos sobre reacciones de muchos tipos que
incluyen prácticamente toda clase de compuestos.