Página principal http://organica1.org
16 Orientación de
la escisión de los epóxidos
En un
anillo de epóxido
hay dos átomos de carbono, cada uno de los cuales
puede sufrir, en
principio, un ataque
nucleofílico. En un
epóxido simétrico, como
el óxido de etileno, ambos carbonos
son equivalentes, por lo que el
ataque se realiza al
azar en cualquiera
de ellos. En
cambio, en un
epóxido no simétrico
los carbonos no
son equivalentes, por
lo que el
producto obtenido dependerá de
cuál es atacado
de preferencia. ¿Cómo
se explica la
orientación de la
escisión en los
epóxidos?
Resulta que
el punto de ataque preferido depende principalmente de
s la reacción
es con catálisis
ácida o básica.
Consideremos, a modo
de empleo, dos
reacciones del óxido
de isobutileno:
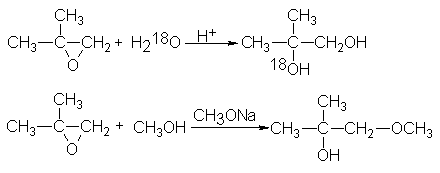
Como sucede
generalmente, en este
caso el nucleófilo
ataca al carbono más sustituido
en la escisión catalizada
por ácidos, y
al menos sustituido, en la escisión catalizada por bases.
Podría
suponerse que se trata
aquí de dos
mecanismos diferentes, SN1 y SN2. Sin
embargo, la evidencia
indica claramente que
ambos son del
tipo SN2: la
ruptura del enlace
carbono-oxígeno y el
ataque del nucleófilo
suceden en una
sola etapa. (No
se trata sólo
de evidencia estereoquímica inversión
completa, sino también
de otros tipos
que no podemos
analizar aquí.) Por
consiguiente, ¿cómo explicar
la orientación diferente
y, en particular
e ataque SN2 en
la posición más impedida
en la catálisis
ácida?
Encontramos este
mismo tipo de orientación en la formación
de halohidrinas, y la explicación
dada allí también
es válida en
este caso. En
el estado de
transición de la
mayoría de las
reacciones SN2, la
ruptura y formación de enlaces
han procedido hasta
un grado más o
menos similar, de
modio que el
cartión no se
ha hecho apreciablemente positivo
o negativo; como
resultado, la reactividad
es determinada por
factores estéricos, en
vez de electrónicos.
Sin embargo, en
la escisión de un
epóxido por catálisis
ácida, el enlace
carbono-oxígeno, débil a
causa de la
tensión angular del anillo de
tres átomos, se
debilita aún más
por la protonación: el grupo
saliente es muy bueno,
un hidroxilo alcohólico
ligeramente básico. En
cambio, el nucleófilo
es malo (agua,
alcohol). En el
estado de transición,
la ruptura de enlace
ha progresado más
que la formación, por
lo que el
carbono ha adquirido
una carga positiva
considerable.
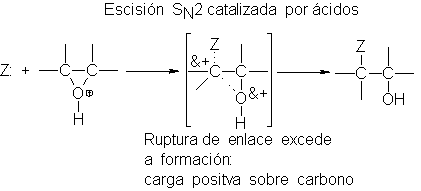
Como tanto
el grupo saliente
como el nucleófilo
están lejos, la
aglomeración es relativamente
poco importante. Al igual
que en la
formación de halohidrinas, la estabilidad
del estado de
transición está determinada
principalmente por factores
electrónicos, no por
estéricos, de modo
que la reacción
tiene un carácter
SN1 considerable.
El ataque ocurre
en el carbono
que mejor acomoda
la carga positiva.
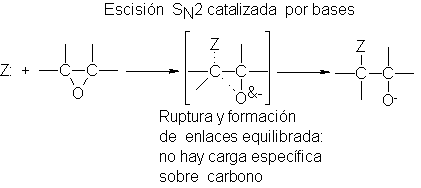
En la
escisión catalizada por
bases, el grupo
saliente es peor
un ox´geno alcoxílico
fuertemente básico y
el nucleófilo es bueno (hidróxido,
alcóxido), por lo que
la ruptura y
formación de enlaces
está más equilibrada,
y la reactividad
se controla en
la forma más
usual, por factores
estéricos. El ataque se
efectúa en el
carbono menos impedido.
Consideremos un
aspecto adicional. Hemos
visto la adición de
reactivos no simétricos, en dos
etapas, donde el
primer paso lo constituye
el ataque con
halógeno positivo: formación
de halohidrinas y
adición heterolítica del IN3 y BrN3. Allí
vimos que la
orientación es similar a
la escisión de
epóxidos catalizada por
ácidos (no por bases):
la que cabría
esperar si se
formara como intermediario
un carbocatión. Por
ejemplo, la fórmula de
la propilén clorhidrina
es CH3CHOHCH2CI; el
reactivo IN3 se
adiciona a alquenos
terminales, produciendo RCH(N3)CH2I. La
estereoquímica es exclusivamente ant
lo que indica que
en estas reacciones
el intermediario no
es un catión
abierto, sino un ion
halogenonio; la escisión
del anillo involucra
el ataque del
nucleófilo (H2O o N3-) al
carbono más impedido,
lo cual no
es sorprendente en
vista de lo
que acabamos de
estudiar sobre los
epóxidos. El anillo
de halogenonio es
aún menos estable
que un epóxido
protonado, por lo
que la ruptura
del enlace es más
fácil; la escisión tiene
mucho carácter SN1 y
se efectúa en
el carbono que
mejor acomoda la
carga positiva.
(Consideremos tambén la orientación
de la solvomercuración, en
la que el
intermediario es un ion
mercurinio cíclico.)