Página
principal http://organica1.org
12
Mecanismo de la hidroboración
Gran parte
de la utilidad de la hidroboración-oxidación radica en la orientación
<<poco común>> de la hidratación. El –OH simplemente pasa a ocupar
la posición del boro en el alquilborano intermediario, por lo que el producto final refleja la orientación del
paso de la hidroboración. ¿Es realmente <<extraordinaria>> esta
orientación?
La
orientación parece inusual porque el hidrógeno se agrega al extremo opuesto del
doble enlace, con respecto al que se une normalmente en una adición
electrofílica común. Sin embargo, la idea fundamental de la adición
electrofílica es que la parte electrófila
del reactivo la parte ácida se une en
forma tal, haciendo uso de los electrones n, que el carbono que pierde estos
electornes sea el que mejor soporte esta pérdida. En la adición de HZ al
propileno, por ejmeplo, el protón se liga al C-1; de esta manera se desarrolla
la carga positiva en el C-2, donde puede ser dispersada mediante el grupo
metilo. Se forma así un carboactión
secundario, en lugar de uno primario.

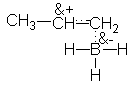 ¿Cuál es el centro de la acidez en el BH3? No cabe
duda que es el boro, con sólo seis
electrones. No debe extrañar que el boro
busque los elctrones n del doble
enlace, comenzando así a unirse al carbono. Al
hacerlo, se liga
de modo tal que
pueda desarrollarse la carga
positiva sobre el
carbono que mejor la
acomode. Así,
¿Cuál es el centro de la acidez en el BH3? No cabe
duda que es el boro, con sólo seis
electrones. No debe extrañar que el boro
busque los elctrones n del doble
enlace, comenzando así a unirse al carbono. Al
hacerlo, se liga
de modo tal que
pueda desarrollarse la carga
positiva sobre el
carbono que mejor la
acomode. Así,
Sin embargo,
a diferencia de la adición eletrofílica
común, la reacción no genera un carbocatión.
A medida que se alcanza el
estado de transición, el carbono
que está perdiendo sus electornes n va
haciéndose progresivamente
más a´cido; el
boro deficiente en
electrones es ácido, pero también lo es el carbono deficiente.
A poca distancia se encuentra un
átomo de hidrógeno unido al boro por un par de electornes. El carbono comienza a tomar ese hidrógeno, con su par de
electrones; a medida que recupera
los elctrones n, el boro
se hace progresivamente propenso
a liberarse de ese
hidrógeno. Tanto el
boro como el hidrógeno
se unen a carbonos del
doble enlace en el mismo
estado de transición:

Debido a
la naturaleza básica de
los alquenos y
a la ácida
del BH3, la principal
fuerza impulsora de la reacción seguramente es la unión
del boro al
carbono. En el estado
de transición, la unión
del boro al C-1 ha procedido más
allá que la del hidrógeno
al C-2, de modo que la pérdida de
electrones (n) por el C-2 en favor
de la unión C-1-B,
excede la ganancia de electrones
del hidrógeno, y así el C-2, el carbono que mejor
puede acomodar la carga,
se ha cargado positivamente.
Por
razones teóricas, se ha postulado que el paso
recién descrito debe seguir a
otro preliminar en el cual
el boro se une a
los dos carbonos
o quizás a los electrones n.
Así, la
orientación de la adición en la hidroboiración es controlada fundamentalmente
de la misma forma que la adición electrofílica
de dos etapas. El hidrógeno se
une a extremos opuestos del doble enlace en las dos reacciones porque lo hace
sin electrones en un caso (en forma de un protón,
un ácido), y con electrones, en el otro (como un ion hidruro, una base).
Debido al
tratamiento de ácidos y bases en el sentido de Lowry-Bronsted, se tiende a
pesar en el hidrógeno principalmente en función de su carácter protónico. Sin embargo, su carácter de hidruro tiene un sentido más real: por ejemplo, el
hidruro de litio sólido tiene un retículo cristalino formado por Li+ y H-;
en cambio, el químico orgánico nunca se enfrenta a un protón desnudo no
solvatado.
Ya nos hemos
familiarizado con la transferencia sencilla de hidruro de un carbono a otro:
dentro de una misma molécula (desplazamiento de hidruro en transposiciones) y
entre moléculas (separación por un carbocatión,). Más adelante veremos un
conjunto de agentes reductores notablemente versátiles (hidruros, como litio y aluminio, LiAlH4, y borohidruro
de sodio, NaBH4) que actúan por transferencia de iones hidruro a
moléculas orgánicas.
La
orientación de la hidroboración se ve afectada,
además de por el factor polar
recién descrito, por uno estérico: la unión de la parte del boro del
reactivo (no sólo -BH2, sino –BHR y –BR2, que son más grandes) es facilitada por el carbono
menos impedido del doble enlace. En general, éste daría la misma orientación
que la aportada por el factor polar por sí solo, aunque no es fácil decidir
cuál de los factores es el que controla. Podemos esperar, no obstante, lo
siguiente: cuanto más voluminosos sean los sustituyentes en el alqueno, mayor
será la importancia del factor
estérico; cuanto más potentes sean los sustituyentes como atractores o
liberadores de electrones, más importante será el factor polar.
a