Alquenos II.
(Ir a página principal http://organica1.org/ )
Adición
electrofílica y de radicales libres
Adición electrofílica y de
radicales libres
2 Reacciones en el doble enlace
carbono-carbono. Adición
3 Hidrogenación. Calor de
hidrogenación
4 Calor de hidrogenación y
estabilidad de los alquenos
6 Adición de bromuro de
hidrógeno. Efecto peróxido
8 Adición de agua. Hidratación
9 Adición electrofílica:
mecanismo
10 Adición electrofílica:
transposiciones
11 Adición electrofílica:
ausencia de intercambio de hidrógeno
12 Adición electrofílica:
orientación y reactividad
14 Mecanismos de la adición de
halógenos
15 Formación de halohidrinas:
adición de los elementos de los ácidos hipohalogenosos
16 Adición de alquenos.
Dimerización
17 Adición de alcanos.
Alquilación
18 Adición de radicales libres.
19 Orientación de la adición de
radicales libres.
20 Otras adiciones de radicales
libres
21 Polimerización de alquenos
por medio de radicales libres
22 Hidroxilación. Formación de
1,2-dioles
.23 Escisión: determinación de
la estructura por degradación. Ozonólisis
1
Reacciones de los alquenos
Como ya
hemos visto, el rasgo característico de la estructura de un alqueno es el doble
enlace carbono-carbono. Este es, por tanto, el grupo funcional de los alquenos, y como tal determina las
reacciones características que sufren estos hidrocarburos.
Las
reacciones son de dos tipos.
(a)
Primero,
las que se verifican en el propio doble enlace y, al hacerlo, lo destruyen.
Estas son las reacciones que estudiaremos
en este capítulo.
(b)
Segundo,
las que suceden, no en el doble enlace, sino en ciertas posiciones que guardan
relaciones especiales con el doble enlace. Aparentemente, el doble enlace no
está involucrado, ya que aparece intacto en el producto. Sin embargo, tiene una
participación esencial, aunque oculta,
en la reacción: determina la velocidad y el mecanismo de la reacción, e incluso
si ésta tiene lugar.
2
Reacciones en el doble enlace carbono-carbono. Adición
¿Qué
tipo de reacción se puede esperar del doble enlace carbono-carbono? El doble
enlace consiste en un enlace s fuerte, y en otro p débil; en consecuencia, es de esperar que la
reacción implique la ruptura de este enlace más débil, lo que resulta correcto;
las reacciones características del doble enlace del tipo:
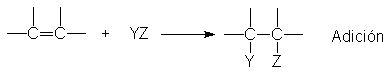
donde
se rompe el enlace p, formándose en su lugar dos
enlaces s fuertes.
Una reacción en la que se combinan dos
moléculas para producir una sola, se llama reacción
de adición. El reactivo simplemente se agrega a la molécula orgánica, en
contraposición a la reacción de sustitución, en la que una parte del reactivo
es sustituido por una porción de la molécula orgánica. Los procesos de adición
se limitan necesariamente a compuestos que tienen átomos que comparten más de un par de electrones; es decir,
sustancias que contienen átomos unidos con enlaces múltiples. La adición es
formalmente lo opuesto a la de la eliminación: así como la eliminación genera
un enlace múltiple, la adición lo destruye.
¿Qué
tipo de reactivo puede unirse al doble enlace carbono-carbono? En la estructura
del enlace hay una nube de electrones p por encima y por debajo del plano de los átomos
(véase Fig. 1). Estos electrones están menos implicados que los s en mantener unidos los núcleos de carbono;
en consecuencia, ellos mismos están más sueltos. Estos electrones n sueltos
están especialmente disponibles para
reactivos que buscan electrones. No es de sorprender, por tanto, que en muchas
de sus reacciones del doble enlace carbono-carbono sirva de fuente electrónica; es decir, actúa
como base. Los compuestos con los
cuales reacciona, son los deficientes en electrones; es decir, que son ácidos. Estos reactivos ácidos que
buscan un par de electrones se llaman reactivos
electrofílicos (del griego, amante de electrones). La reacción típica de un alqueno es la adición electrofílica o, en
otras palabras, la adición de reactivos ácidos.
Fig. 1 Doble enlace carbono-carbono: la unión
n es una fuente de electrones.
Los
reactivos de otro tipo, los radicales libres,
buscan electrones o, más bien, buscan un electrón. Así pues, los alquenos
también sufren adición de radicales
libres.
Los
alquenos no sólo contienen el doble enlace, sino también grupos alquilo, que
tienen esencialmente la estructura de los
alcanos. En consecuencia, aparte de las reacciones de libres
característica de los alcanos. Más adelante se resumirán las reacciones más
importantes de adición y sustitución, que se estudiarán en detalle en las
siguientes secciones: en este capítulo y posteriores.
Hay
reactivos que se pueden adicionar como ácidos, o como radicales libres, y con
resultados notablemente diferentes; algunos pueden agregarse al doble enlace y
provocar sustitución. Veremos que, según sean las condiciones que elijamos,
podemos conducir estos reactivos al proceso deseado: electrofílico o de
radicales libres, adición o sustitución.
Los
grupos alquilo unidos a los carbonos del doble enlace modifican las reacciones
de éste, que, a su vez, altera las del grupo alquilo. Veremos cuáles son estas
modificaciones y, donde sea posible, cómo se pueden explicar.
En los
capítulos 9 y 20 estudiaremos la estereoquímica de estas reacciones de adición,
tanto por motivos prácticos, conocer lo que probablemente obtendremos en una
síntesis, como por lo que nos puede revelar sobre la forma en que suceden estas
reacciones.
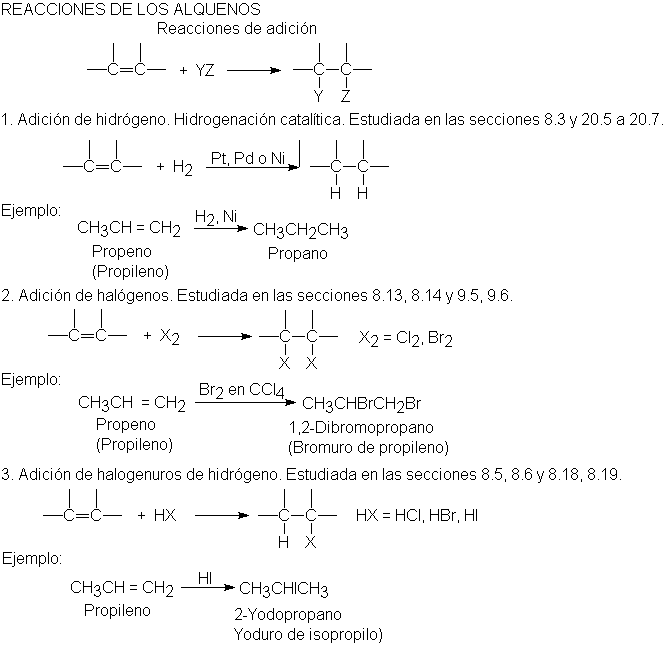

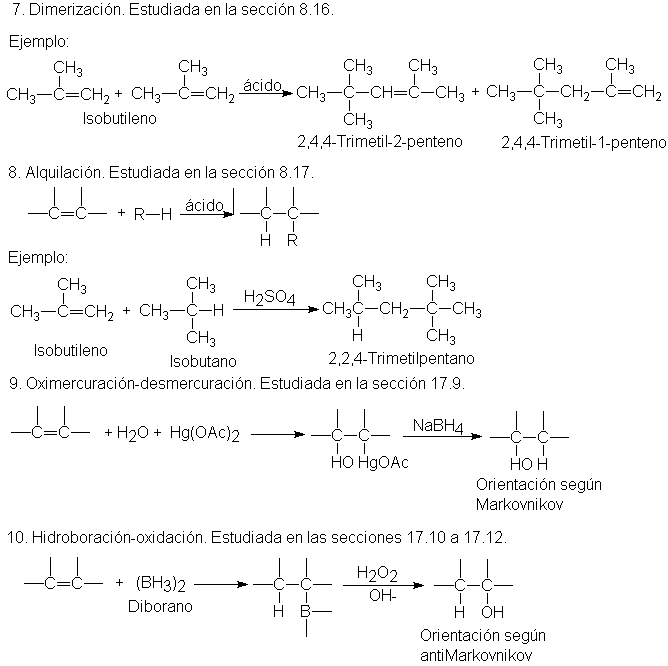

3
Hidrogenación. Calor de hidrogenación
Hemos
establecido que la hidrogenación es el modo más útil de preparar alcanos.
No se
limita a la síntesis de alcanos, sino que es método general para convertir un
doble enlace carbono-carbono en uno simple, prácticamente en todo tipo de
compuestos. Empleando el mismo equipo, igual catalizador y condiciones muy
parecidas, podemos convertir un alqueno en un alcano, un alcohol no saturado en
uno saturado, o un éster no saturado en uno saturado. Modificando el catalizador
y las condiciones, podemos hidrogenar selectivamente un enlace múltiple sin
tocar en la misma molécula un doble enlace carbono-carbono, pero no uno
carbono-oxígeno; uno triple, pero no uno doble; incluso un doble enlace
carbono-carbono, pero no otro. Como veremos, podemos convertir incluso un
compuesto no saturado ópticamente inactivo en un producto activo.
La
hidrogenación es de dos tipos generales: (a) heterogénea (dos fases) y (b) homogénea
(una fase). En ambos casos, el catalizador provoca la adición de hidrógeno
molecular, H2, al doble enlace.
·
La hidrogenación heterogénea es el método clásico, todavía
muy utilizado. El catalizador es algún metal dividido finamente, por lo común
platino, paladio o níquel. Se agita una solución del alqueno bajo una ligera
presión de hidrógeno gas en presencia de una pequeña cantidad del catalizador.
La reacción es rápida y suave y, una vez completa, simplemente se filtra la
solución del producto saturado del catalizador insoluble.
·
La hidrogenación homogénea, mucho más moderna, ofrece una
flexibilidad imposible de alcanzar con los catalizadores antiguos. Mediante
modificaciones en los catalizadores, puede
llevarse a cabo la hidrogenación con una selectividad sin precedente.
Los catalizadores son complejos orgánicos de metales de transición, como rodio
o iridio: por ejemplo, el catalizador de
Wilkinson. Son solubles en disolventes orgánicos y la hidrogenación se
efectúa así en una sola fase, la solución. Lo inconveniente del método está en
la dificultad de separación del catalizador y el producto una vez terminada la
reacción. Sin embargo, se están desarrollando métodos para evitar esta
dificultad: el catalizador se une incorporado químicamente a un polímero sólido
insoluble (una molécula gigante), lo que permite una filtración fácil al final de la reacción. De esta
manera, la hidrogenación homogénea se convierte en heterogénea, pero el modo de
acción parece permanecer igual. En el capítulo 20 estudiaremos con algún
detalle estos catalizadores: su estructura, cómo trabajan y, en particular,
cómo permiten el control estereoquímico de la hidrogenación y de muchas otras
reacciones.
·
Dado
que la reacción suele ser cuantitativa y es fácil determinar el volumen de hidrógeno consumido, frecuentemente se
usa la hidrogenación como herramienta analítica; por ejemplo, puede indicar el número de dobles enlaces de un
compuesto.
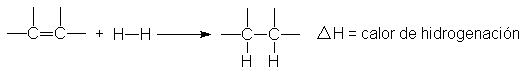
La
hidrogenación es exotérmica: los dos enlaces o (C-H) que se forman son, en
conjunto, más firmes que los enlaces o (H-H) y n que se rompen. La cantidad de calor desprendida al
hidrogenar un mol de un compuesto se llama calor de hidrogenación; es
simplemente el H de la reacción, pero no se incluye el signo menos. El
calor de hidrogenación de casi todo alqueno se aproxima bastante a un valor de
30 kcal por cada doble enlace del compuesto (véase Tabla 8.1)
Tabla 1 CALORES DE HIDROGENACION DE LOS
ALQUENOS
|
Alqueno |
Calor de hidrogenación kcal/mol |
|
Etileno |
32.8 |
|
Propileno |
30.1 |
|
1-Buteno |
30.3 |
|
1-Penteno |
30.1 |
|
1-Hepteno |
30.1 |
|
3-Metil-1-buteno |
30.3 |
|
3,3-Dimetil-1-buteno |
30.3 |
|
4,4-Dimetil-1-penteno |
29.5 |
|
Cis-2-Buteno |
28.6 |
|
Trans-2-Buteno |
27.6 |
|
Isobutileno |
28.4 |
|
Cis-2-Penteno |
28.6 |
|
Trans-2-Penteno |
27.6 |
|
2-Metil-1-buteno |
28.5 |
|
2,3-Dimetil-1-buteno |
28.0 |
|
2-Metil-2-buteno |
26.9 |
|
2,3-Dimetil-2-buteno |
26.6 |
La hidrogenación
procede a una velocidad despreciable en ausencia de un catalizador, aun a
temperaturas elevadas, a pesar de ser
una reacción exotérmica, por lo que el proceso no catalizado debe tener una
energía de activación muy alta. La función del catalizador es reducir la
energía de activación (Eact), de modo que la reacción pueda proceder
rápidamente a temperatura ambiente. Por
supuesto, el catalizador no afecta al cambio neto de energía del proceso
total: sólo rebaja la colina energética entre los reactivos y los productos
(véase Figura 8.2).
Un
catalizador rebaja la Eact permitiendo que la reacción proceda de un
modo distinto, es decir, por medio de un mecanismo diferente. En ese caso, los
reactivos se adsorben en la enorme superficie del metal sólido dividido
finamente, o se unen temporalmente a un ión metálico soluble.En estas
condiciones, la reacción es muy distinta a la que tendría lugar en otro caso.
Se cree, por ejemplo, que la superficie catalítica rompe el enlace n del
alqueno antes de la reacción con el hidrógeno. En la hidrogenación homogénea,
el complejo del ión metálico rompe el enlace hidrógeno-hidrógeno y transfiere
estos átomos, de uno en uno, al doble enlace.
Al
disminuir la colina energética, también se reduce la energía de activación de
la reacción inversa, con lo que aumenta la velcoidad de la deshidrogenación. Es de esperar, entonces, que los
catalizadores metálicos, platino, paladio y níquel, sirvan como
deshidrogenantes en condiciones apropiadas, y efectivamente éste es el caso (Sec.
34.7). Estamos familiarizados con el hecho de que un catalizador acelera una
reacción, pero no desplaza la posición de equilibrio; por supuesto, esto se
debe a que acelera tanto la reacción directa como la inversa.
Al
igual que la hidrogenación, la adición de otros reactivos al doble enlace es
generalmente exotérmica. Casi siempre, la energía consumida en la ruptura de
lso enlaces Y-Z y n es menor que la liberada en la formación de los enlaces C-Y
y C-Z.
4
Calor de hidrogenación y estabilidad de los alquenos
A
menudo, los calentadores de hidrogenación pueden brindarnos información valiosa
acerca de las estabilidades relativas de compuestos no saturados. En los
2-butenos isómeros, por ejemplo, el cis tiene un calor de hidrogenación de 28.6
kcal, y el trans, de 27.6 kcal. Ambas reacciones consumen un mol de hidrógeno y
dan el mismo producto, n-butano.Por tanto, si el isómero trans libera 1 kcal
menos de energía que el cis, sólo puede significar que contiene 1 kcal menos de
energía; en otras palabras, el isómero trans es más estable que el cisen 1 kcal
(véase Fig. 8.3). De modo análogo, el trans-2-penteno (calor de hidrogenación =
27.6 kcal) debe ser más estable en 1.0 que el cis-2-penteno (calor de
hidrogenación = 28.6 kcal).
De los
alquenos simples disustituidos, el isómero trans generalmente es el más
estable;
los dos
sustituyentes mayores están más separados que en el isómero cis; hay menos
impedimento y menor tensión de Van der Waals (Sec. 7.6).
Los
calores de hidrogenación indican que la estabilidad de un alqueno también
depende de la ubicación del doble enlace. Son característicos los ejemplos
siguientes:
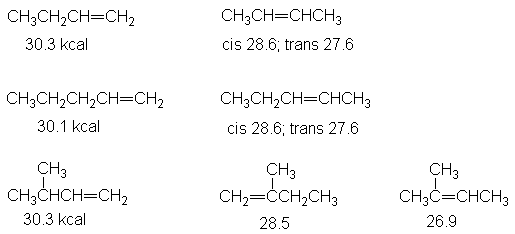
Cada
grupo de alquenos isómeros da el mismo alcano. En consecuencia, las diferencias
en calores de hidrogenación deben proceder de diferencias en estabilidades. En
cada caso, se observa que cuanto mayor es el número de grupos alquilo unidos a
los carbonos del doble enlace, más estable es el alqueno.
![]()
Hemos
visto antes el papel importante que desempeña la estabilidad de los alquenos en
la orientación y reactividad de las reacciones de eliminación.
5 Adición de halogenuros de hidrógeno. Regla de Markovnikov.
Reacciones
regioselectivas
El
cloruro, bromuro o yoduro de hidrógeno convierten un alqueno en el halogenuro
de alquilo correspondiente.
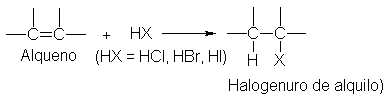
Frecuentemente,
la reacción se realiza haciendo pasar directamente por el alqueno halogenuro de
hidrógeno gaseoso y seco. A vecces, se emplea el ácido acético, que es un
disolvente moderadamente polar, ya que disuelve tanto el halogenuro de hidrógeno polar como el alqueno no polar.
Generalmente no se emplean las conocidas soluciones acuosas de los halogenuros
de hidrógeno; en parte, esto se debe a que así se avita la adición de agua al
alqueno .
El
etileno se convierte así en un halogenuro de etilo, uniéndose el hidrógeno a
uno de los carbonos del doble enlace, y
el halógeno, al otro.
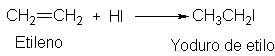
El
propileno podría dar dos productos: el halogenuro de n-propilo o el halogenuro
de isopropilo, dependiendo de la orientación de la adición; es decir,
dependiendo de a qué átomos de carbono se unen al hidrógeno y el halógeno. De
hecho, sólo se forma el halogenuro de isopropilo.
Del
mismo modo, el isobutileno podría dar dos productos: el halogenuro de isobutilo
o del t-butilo; en este caso, la orientación de la adición es tal que sólo se
forma el halogenuro de t-butilo.
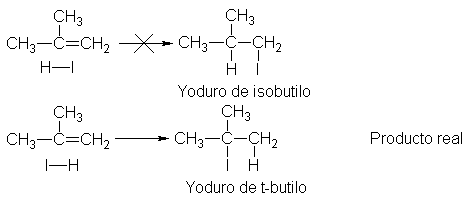
Así, en
la adición de un reactivo YZ a un alqueno, la orientación depende de qué
carbonos del doble enlace aceptan a Y y de cuáles aceptan a Z.
Al
examinar adiciones como éstas, el químico ruso Vladimir Markovnikov
(Universidad de Kazán) comprobó que cuando existe la posibilidad de formación
de dso productos isómeros, generalmente predomina uno. En 1869, señaló que la orientación de la adición sigue un
esquema que puede resumirse de la manera siguiente: En la adición iónica de un
ácido al doble enlace de un alqueno, el hidrógeno de aquél se une al átomo de
carbono que ya tiene el mayor número de hidrógenos. Este enunciado se conoce
generalmente como regla de Markovnikov. Puede describirse también: Al que
tiene, le será dado o El que tiene, recibirá.
En la
adición al propileno, por tanto, observamos que el hidrógeno se une al carbono
que tiene dos hidrógenos y no al que tiene uno; en la adición al isobutileno el
hidrogeno va al que tiene dos hidrógenos y no al que no tiene ninguno.
Empleando
la regla de Markovnikov, podemos predecir correctamente el producto principal
de muchas reacciones. Por ejemplo:
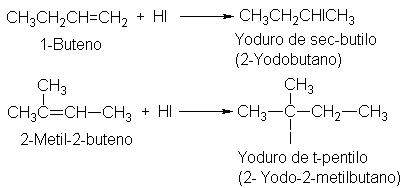
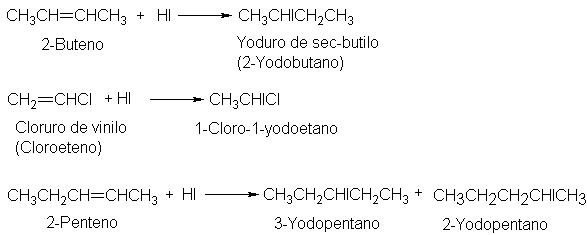
En el
2-penteno cada uno de los carbonos del doble enlace tiene un hidrógeno, de modo
que, según la regla, no debe predominar ninguno de los dos productos. De nuevo,
el pronóstico es esencialmente correcto, pues de hecho se obtienen cantidades
casi iguales de ambos isómeros.
Los
ejemplos implican la adición del yoduro de hidrógeno; resultados exactamente
análogos se obtienen con la adición de clorurp de hidrógeno y, salvo
condiciones especiales indicadas en la siguiente sección, también con la de
bromuro de hidrógeno.
Considerando
la orientación, las reacciones que dan exclusiva o casi exclusivamente uno de
varios productos isómeros posibles, se denominan regioselectivas (del latín:
regio, dirección).
6
Adición de bromuro de hidrógeno. Efecto peróxido
Las
adiciones de cloruro y yoduro de hidrógeno siguen la regla de Markovnikov.
Hasta 1933, la situación con respecto al bromuro de hidrógeno era
extremadamente confusa.
Algunos
investigadores habían informado que la adición de bromuro de hidrógeno a un
alqueno determinado daba un producto de acuerdo con la regla de
Markovnikov; otros, que el producto
obtenido era contrario a la regla, y aún otros, que resultaban una mezcla de
ambos. También había publicaciones en las que se decía que el producto obtenido
dependía de la presencia o ausencia de agua, o de la luz, o de ciertos
halogenuros metálicos; se decía que el
producto obtenido dependía del disolvente empleado o de la naturaleza de la
superficie de la cubeta de reacción.
En
1933, M. S. Kharasch y F.R. Mayo (Universidad
de Chicago) dieron respuesta a esta controversia al descubrir que la
orientación de la adición del bromuro de hidrógeno al doble enlace
carbono-carbono está determinada exclusivamente por la presencia o ausencia de
peróxidos.
Los
peróxidos orgánicos son compuestos que contienen la unión O-O-. Generalmente se
encuentran como impurezas de muchas sustancias orgánicas en cantidades muy pequeñas formadas lentamente por la
acción del oxígeno. Ciertos peróxidos se sintetizan deliberadamente y se
emplean como reactivos: peróxidos de
t-butilo (Cap. 3, Problema 20), o peróxido de benzoilo (C6H5COO)2, por ejemplo.
Kharasch
y Mayo indicaron que si se
excluyen con cuidado los peróxidos del
sistema de reacción o si se agregan ciertos inhibidores por ejemplo,
hidroquinona (Sec.31.9) o difenilamina (Sec. 26.3)-, la adición del HBr a los
alquenos sigue la regla de Markovnikov.
Por
otra parte, si no se excluyen los peróxidos, o si se agregan deliberadamente al
sistema de reacción, el HBr se adiciona a los alquenos en dirección exactamente
opuesta.
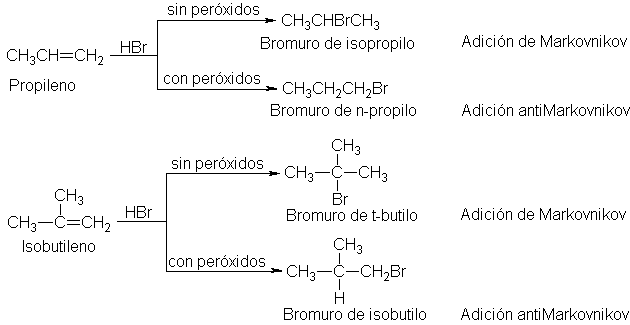
Esta
inversión en la orientación, causada por los peróxidos, seconoce como el efecto
peróxido. De las reacciones que estamos estudiando, solamente la adición de
bromuro de hidrógeno presenta tal efecto. La presencia o ausencia de los
peróxidos no afecta a la orientación de la adición del cloruro y yoduro de
hidrógeno, ácido sulfúrico, agua, etc. Más adelante veremos que tanto la regla
de Markovnikov como el efecto peróxido
tienen una explicación sencilla y consecuentemente con la química que hemos
analizado hasta este momento.
7
Adición del ácido sulfúrico
Los
alquenos reaccionan con el ácido sulfúrico concentrado y frío formando
compuestos de fórmula general ROSO3H, conocidos como sulfatos ácidos de alquilo.
Estos productos se generan por adición de hidrógeno a un cabrono del doble
enlace, y de ión bisulfato, al otro.
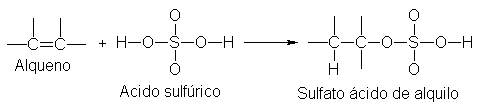
Al
igual que los sulfonatos de alquilo estos compuestos son ésteres: ésteres del
ácido sulfúrico, al igual que los sulfonatos de alquilo son ésteres de los
ácidos sulfónicos.
La
reacción se lleva a cabo simplemente juntando los reactivos: se burbujea un
alqueno gaseoso a través del ácido y se agita un alqueno líquido con él. Puesto
que los sulfonatos ácidos de alquilo son solubles en ácido sulfúrico, resulta
una solución clara. Los sulfonatos
ácidos de alquilo son sólidos delicuescentes y es difícil aislarlos. La
concentración del a´cido sulfúrico requerido para la reacción depende del alqueno específico, como ilustran lso siguientes
ejemplos; explicaremos esto más adelante.
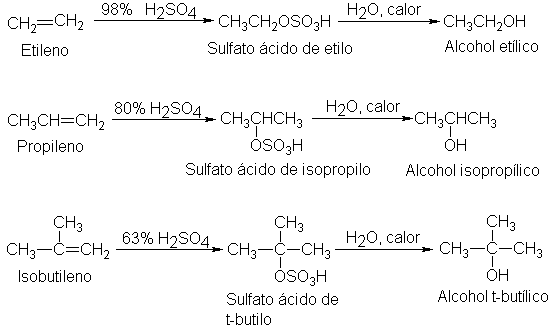
Si se
diluye la solución sulfúrica del sulfato ácido de alquilo con agua, y se
calienta, se obtiene un alcohol con el mismo grupoalquilo que elsulfato
original. El éster ha sido descompuesto por el agua para formar el alcohol y
a´cido sulfúrico, y se diceque se ha hidrolizado. Esta serie de reacciones proporciona una vía para separar alcoholes: con este propósito generalmente
se efectúa la adición del a´cido sulfúrico a los alquenos. El anterior es un
método excelente para la manufactura de alcoholes en gran escala, puesto que
los alquenos son fáciles de obtener por
el cracking del petróleo. Ciertos alcoholes no pueden obtenerse por este métodoporque la adición del ácido sulfúrico sigue
la regla de Markovnikov.
Por
ejemplo, pueden hacerse alcohol isopropílico, pero no el n-propílico; puede
obtenerse el t-butílico, pero no el isobutílico.
El
hecho de que los alquenos se disuelven en ácido sulfúrico concentrado y frío
para formar sulfatos sirve para purificar
ciertos tipos de compuestos. Los alcanos y los halogenuros de alquilo, por ejemplo, insolubles en ácido
sulfúrico, pueden liberarse de los alquenos que tengan impurezas por medio de
un lavado con el ácido: un alcano gaseoso se burbujea a través de varios
frascos con ácido sulfúrico, y se agita un alcano líquido con ácido sulfúrico
en un ambudo de decantación.
8
Adición de agua. Hidratación
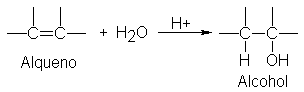 El agua se adiciona a los
alquenos más reactivos, en presencia de ácidos, para dar alcoholes. Puesto que
esta adición también sigue la regla de Markovnikov, se obtienen los mismos
alcoholes que por la síntesis de dos etapas recién descrita. Esta hidratación
de alquenos,
El agua se adiciona a los
alquenos más reactivos, en presencia de ácidos, para dar alcoholes. Puesto que
esta adición también sigue la regla de Markovnikov, se obtienen los mismos
alcoholes que por la síntesis de dos etapas recién descrita. Esta hidratación
de alquenos,
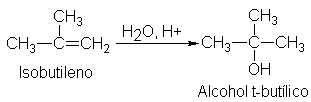 ya sea directa o por la vía de
los sulfatos ácidos de alquilo, es la principal fuente industrial de aquellos
alcoholes más simples cuya formación concuerda con la regla de Markovnikov.
ya sea directa o por la vía de
los sulfatos ácidos de alquilo, es la principal fuente industrial de aquellos
alcoholes más simples cuya formación concuerda con la regla de Markovnikov.
9
Adición electrofílica: mecanismo
Antes
de considerar otras reacciones de los alquenos, es útil examinar el mecanismo
de algunas de las reacciones que hemos tratado. Una vez hecho esto, volveremos
al estudio sistemático de las reacciones de los alquenos, para entenderlas
mejor en fusión de las primeras.
La
adición del reactivo ácido, HZ, procede
en dos pasos:
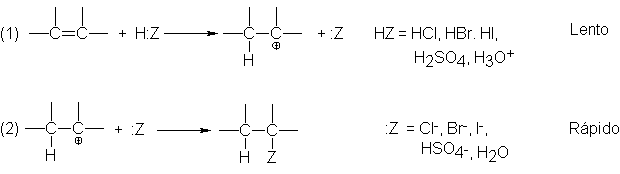
El paso
(1) implica la tranferencia del ión hidrógeno de :Z al alqueno para formar un
carbocatión: la transferencia de un protón de una base a otra.
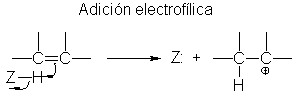
El paso
(2) es la unión del carbocatión con la base :Z.
Veamos
lo que sucede en el paso (1), centrado nuestra atención en el HZ y los dos
carbonos con doble enlace del alqueno. Se transfiere hidrógeno en forma de
protón es decir, sin sus electrones, que se dejan en la base :Z-. Para
establecer el enalce con el hidrógeno, el carbono utiliza lso electrones n que
antes compartía con el otro carbono, que
queda ahora con un sexteto de electrones solamente, y se convierte en el
carbono deficiente en electrones de un carbocatión.
El paso
(1) es la etapa difícil y su velocidad controla en gran medida o enteramente la
velocidad total de la adición. Este paso implica un ataque por medio de un
reactivo ácido buscador de electrones es decir, un reactivo electrofílico-, por
lo que el proceso se denomina adición electrofílica. El electrófilo, como se
indica aquí, no debe necesariamente ser un ácido de Lowry-Bronsted que
transfiere un protón, puede ser, como
veremos, casi cualquier tipo de
molécula deficiente en electrones (ácidos de Lewis).
El
mecanismo general puede ilustrarse con ejemplos específicos: adición de cloruro
de hidrógeno,
Observamos
que el carbocatión se combina con el agua no para dar el alcohol, sino el
alcohol protonado, que en una reación posterior cede un ión hidrógeno a otra
base, generando el alcohol. Según se aprecia, esta serie de reacciones es
exactamente la inversa de la propuesta para la deshidratación de los alcoholes
(Sec. 7.25) En la deshidratación, los equilibrios se desplazan en favor del
alqueno, debido principalmente a que éste se elimina de la mezcla de reacción
por destilación; en la hidratación, lso
equilibrios se desplazan hacia la fromación del alcohol, debido en aprte a la
elevada concentración de agua.
¿Cuál
es la evidencia para este mecanismo? La siguiente:
(a)
La
velocidad de la reacción depende de la concentración del alqueno y del reactivo
HZ.
(b)
La reacción requiere un reactivo ácido.
(c)
Donde la estructura lo permite, la reacción
va acompañada de transposiciones.
(d)
El alqueno no sufre intercambio apreciable de
hidrógeno.
Además,
el mecanismo concuerda con:
(e) la
orientación de la adición, y
(f) las
reactividades relativas de los alquenos.
Examinemos
esta evidencia.
Primero,
(a) la velocidad de la reacción depende de la concentración del alqueno y del
reactivo HZ. Este hecho es, por
supuesto, concordante con un mecanismo que comienza con la reacción
entre estos dos reactivos.
Segundo,
(b) la reacción requiere un reactivo ácido. De acuerdo con el mecanismo, el
primer paso en todas estas reacciones es la trasnferencia de un protón al
alqueno. Esto concuerda con el hecho de que todos estos reactivos, excepto el
agua, son ácidos fuertes en el sentido de Lowry-Bronsted; es decir, pueden
transferir protones con facilidad. La excepción, el agua, requiere la presencia
del ácido fuerte añadido para que se efectúe la reacción. Un alqueno es una
base débil y acepta protones en un grado significativo solamente de ácidos
fuertes.
Incluso
el familiar ácido acético (CH3COOH) no es suficientemente fuerte como para
reaccionar por sí solo con alquenos. Sin emabrgo, cuando se usa ácido acético
como disolvente para la adición de HCI, se obtiene, junto con el cloruro de
alquilo, una cantidad menor del acetato
de alquilo (CH3COOR). Este no se genera por una reacción de sustitución entre
ácido acético y cloruro de alquilo ya formado por adición; en las condiciones de la reacción, el cloruro de alquilo es inerte
al ácido acético. La interpretación más sencilla de esto es que el ácido fuerte
convierte al alqueno en algo que puede reaccionar con agua o ácido acético.
Según el mecanismo, ese algo es el carbocatión.
10
Adición electrofílica: transposiciones
Donde
la estructura lo permite, (c) la reacción va acompañada de transposiciones. El
producto a veces contiene el grupo Z ligado a un carbono que no estaba unido
mediante un doble enlace en el sustarto; en otras ocasiones, el producto
incluso tiene un esqueleto carbonado distinto del que presentaba el sustrato.
Estos
productos inesperados pueden justificarse sin dificultad por transposiciones de
los carbocationes propuestos como intermediarios. Las transposiciones siguen la
misma pauta que hemos observado en nuestro estudio de los carbocationes en la
sustitución SN1 (Sec. 5.23) y en la eliminación E1 (Secs. 7.21 y 7.25).
La
adición de cloruro de hidrógeno al 3-metil-1-buteno, por ejemplo, no sólo
genera el 2-cloro-3-metilbutano esperado, sino también 2-cloro-2-metilbutano.
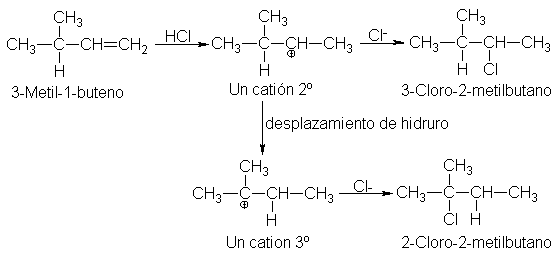
Puesto
que un desplazamiento 1,2 de hidrógeno puede convertir el catión secundario
inicialmente formado en otro terciario más estable, tal transposición va a
ocurrir, derivándose gran parte del producto de este nuevo catión.
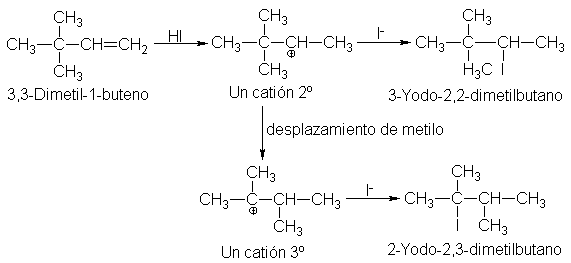 La adición de yoduro de
hidrógeno al 3,3-dimetil-1-buteno no
sólo da el 3-yodo-2,2-dimetilbutano esperado, sino también
2-yodo-2,3-dimetilbutano:
La adición de yoduro de
hidrógeno al 3,3-dimetil-1-buteno no
sólo da el 3-yodo-2,2-dimetilbutano esperado, sino también
2-yodo-2,3-dimetilbutano:
Observamos
otra vez la conversión de un catión secundario en uno terciario, ahora mediante
un desplazamiento 1,2 de un grupo metilo.
El
cambio en el esqueleto carbonado que acompaña este último ejemplo de adición es
idéntico al que acompaña dos reacciones del 3,3-dimetil-2-butanol:
deshidratación (Sec. 7.25), una reacción de eliminación; y la conversión en el
cloruro (Sec. 5.25), una sustitución. Este es un ejemplo particularmente
representativo del tipo de prueba que dio origen a la idea de que estas
reacciones aparentemente no relacionadas proceden por la vía del mismo
intermediario: el carbocatión.
De toda
la evidencia que apoya el mecanismo presentado para la adición electrofílica,
la más poderosa es la existencia de las transposiciones, puesto que apuntan
directamente al fondo del mecanismo: la formación del carbocatión.
11
Adición electrofílica: ausencia de intercambio de hidrógeno
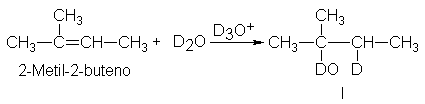 En la adición electrofílica, (d)
el alqueno no sufre un intercambio apreciable de hidrógeno. La adición de D2O
al 2-metil-2-buteno en presencia de D3O+ da el alcohol deuterio I, como era de
esperar.
En la adición electrofílica, (d)
el alqueno no sufre un intercambio apreciable de hidrógeno. La adición de D2O
al 2-metil-2-buteno en presencia de D3O+ da el alcohol deuterio I, como era de
esperar.
Transcurrida
la reacción hasta aproximadamente la mitad, se interrumpe y se aísla el alqueno
no comsumido. El análisis espectrométrico de masas indica la ausencia casi
total de deuterio; es decir, el alqueno no sufre un intercambio apreciable de
hidrógeno o deuterio.
¿Cuál
es la importancia del hallazgo? Esta técnica ya ha sido descrita (Sec. 7.18),
de modo que deberíamos sospechar hacia dónde lleva la solución. Consideremos lo que sucedería si
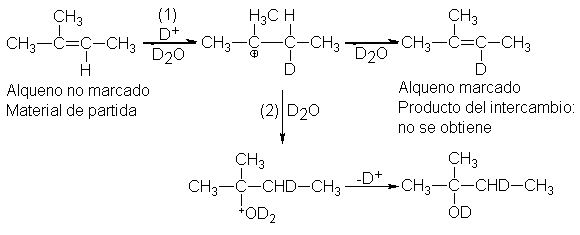
los
carbocationes se formaran rápida y
reversiblemente en el paso (1) y luego, de vez en cuando, se combinaran
lentamente con la base para completar la adición. En tal caso, la mayoría de
los carbocationes perderían hidrógeno
muchas veces para regenerar el alqueno antes de seguir hasta el
producto. Pero en el carbocatión hay dos hidrógenos en el C-3: el protio (H),
que siempre ha estado allí, y el deuterio (D), recientemente adquirido. Al
revertir al alqueno, el carbocatión
perdería con probabilidad similar en realidad, con mayor probabilidad (¿por qué?) tanto protio como
deuterio, por lo que quedaría deuterio en el alqueno. Mientras está en la
solución hasta la mitad de la reacción, la parte no consumida del alqueno intercambiaría
gran parte de su protio por deuterio, y al ser recuperado se hallaría
fuertemente deuterado; lo que es contrario a los hechos.
Esta
evidencia prueba que sí se forman carbocationes y otras evidencias indican que
se generan-, se combinan con la base mucho más velozmente de lo que revierten
al alqueno. Esto significa que el paso (1) es el lento, el determinante de la
velocidad, como muestra el mecanismo de la sección 9. La velocidad de la adición depende principalmente de la
velocidad de formación del carbocatión.
12
Adición electrofílica: orientación y reactividad
El
mecanismo es consecuente con (e) la orientación de la adición de reactivos
ácidos y con (f) el efecto de la estructura sobre las reactividades relativas
de los alquenos.
A
continuación se ilustra la adición del
cloruro de hidrógeno a tres alquenos característicos, indicando los dos pasos
del mecanismo. De acuerdo con la regla de Markovnikov, el propileno da cloruro
de isopropilo; el isobutileno, cloruro de t-butilo, y el 2-metil-2-buteno, cloruro de t-pentilo.
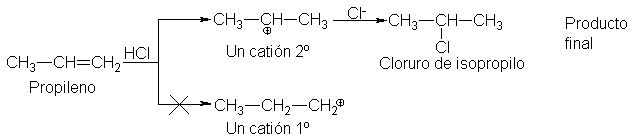
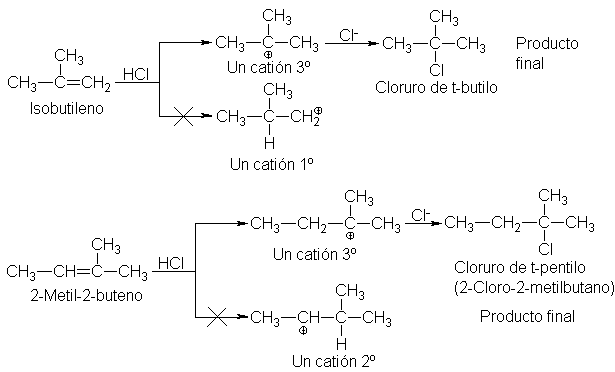
Según
el mecanismo, el hidrógeno del reactivo se adiciona a uno de los dos carbonos
con doble enlace, dando uno u otro de dos carbocationes posibles. Por ejemplo,
si el hidrógeno va al C-2 del propileno, se forma el catión n-propilo; si va al
C-1, se forma el catión isopropilo. Una vez generado, el carbocatión reacciona
velozmente para dar el producto. El halogenuro obtenido depende del carbocatión
que se genre en el primer paso.
El
hecho de que el propileno dé cloruro de isopropilo en lugar de n-propilo,
demuestra que se forma el catión isopropilo en lugar del catión n-propilo es
decir, se forma más rápido.
De esta
manera, en la adición electrofílica, la orientación está determinada por la velocidad relativa de dos
reacciones en competencia: la formación de uno u otro carbocatión.
En cada
uno de los ejemplos expuestos, el producto obtenido demuestra que el primer
paso se forma más velozmente un catión secundario que uno primario, o uno terciario
que uno primario, o uno terciario que uno secundairo. La observación de mcuhos
casos de adición muestra que esta es una regla general: en la adición
electrofílica, la velcoidad deformación de carbocationes sigue la secuencia
Velocidad de formación de carbocationes 3º > 2º > 1º > CH3+
Al
ordenar los carbocationes de acuerdo con la velocidad de su formació a partir
de alquenos, encontramos una vez que
los hemos ordenado según sus estabilidades.
Estabilidad de carbocationes 3º > 2º > 1º > CH3+
Ahora
podemos formular la regla de markovnikov:
la adición electrofílica a un doble enlace carbono-carbono implica la formación intermediaria del
carbocatión más estable.
Al
igual que la regla de Saytzeff esta
reformulación no sólo da una regla de aplicación más general, sino que conduce
al factor realmente operacional.
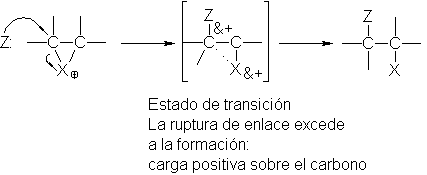
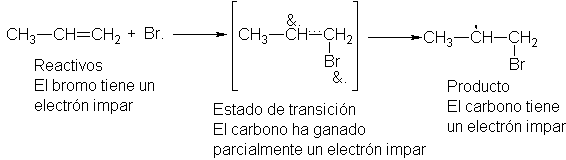
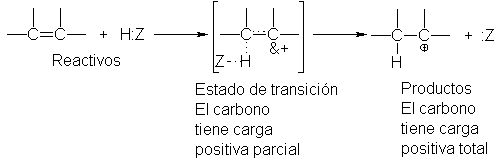 ¿Cómo podemos explicar el hecho de que la velocidad de formación de un carbocatión en la adición electrofílica depende de su
estabilidad? Debemos comparar otra vez
la estructura de los
reactivos con la del estado de
transición. En los reactivos, el hidrógeno está ligado a :Z, y los carbonos del
doble enlace se mantienen unidos no sólo mediante un enalce o, sino también por
un enlace n. En los productos, el hidrógeno está ligado a uno de los carbonos;
se ha roto el enlace n, y el otro carbono ha quedado con un sexteto de
electrones solamente, por lo que tiene carga positiva. En el estado de
transición, el enalce entre hidrógeno y :Z está parcialmente roto, y entre
hidrógeno y carbono está parcialmente establecido. El enlace n está
parcialmente roto, y el carbono ha ganado parcialmente la carga positiva que
llevará en el carbocatión.
¿Cómo podemos explicar el hecho de que la velocidad de formación de un carbocatión en la adición electrofílica depende de su
estabilidad? Debemos comparar otra vez
la estructura de los
reactivos con la del estado de
transición. En los reactivos, el hidrógeno está ligado a :Z, y los carbonos del
doble enlace se mantienen unidos no sólo mediante un enalce o, sino también por
un enlace n. En los productos, el hidrógeno está ligado a uno de los carbonos;
se ha roto el enlace n, y el otro carbono ha quedado con un sexteto de
electrones solamente, por lo que tiene carga positiva. En el estado de
transición, el enalce entre hidrógeno y :Z está parcialmente roto, y entre
hidrógeno y carbono está parcialmente establecido. El enlace n está
parcialmente roto, y el carbono ha ganado parcialmente la carga positiva que
llevará en el carbocatión.
Los
grupos liberados de electrones tienden
a dispersar la carga positiva parcial que se está desarrollando sobre el
carbono, estabilizadoasí el estado de transición. La estabilización del estado
de transición reduce la Eact y permite
una reacción más rápida En la medida en
que se haya roto el enalce n, el grupo orgánico posee el carácter del
carbocatión que va a ser. En otras ocasiones, el mismo factor que estabiliza el
carbocatión, la liberación de electrones, también estabiliza al carbocatión
incipiente en el estado de transición .Nuevamente, vemos que cuanto más estable
es el carbocatión, más velozmente se forma.
Así es
como la adición de un ión hidrógeno al doble enlace depende de la estabilidad
del carbocatión en formación. Como es de esperar, este factor no sólo determina
la orientación de la adición a un alqueno simple, sino también las
reactividades relativas de alquenos diferentes.
Por lo
común, los alquenos muestran el siguiente orden de reactividad hacia la adición
de ácidos:
Reactividad
de los alquenos hacia los ácidos
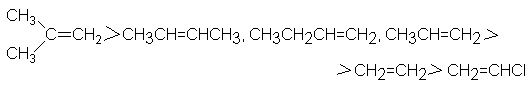
El
isobutileno, que forma un catión terciario, reacciona más velozmente que el
2-buteno, que forma uno secundario. El
1- y el 2-buteno, y el propileno, que forman cationes secundarios, reaccionan
con mayor velocidad que el etileno, que forma un ión primario.
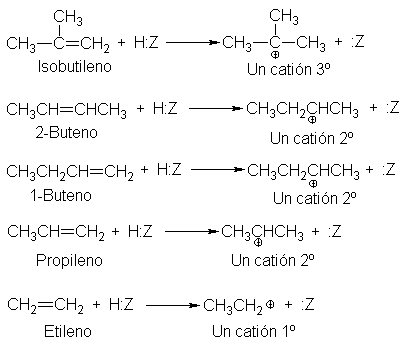
Como
sustituyentes, los halógenos tienden a atraer electrones. Al igual que la
liberación de electrones por los grupos alquilo dispersa la carga positiva y
estabiliza al carbocatión, la atracción de electrones por parte de los
halógenos intensifica dicha carga y resta estabilidad al carbocatión. Hemos
visto que esta atracción de electrones retarda la formación de carbocationes en
la heterólisis; de igual forma, hace más lenta su formación en la adición
electrofílica. Por ejemplo, el cloruro de vinilo, CH2=CHCI, es menor reactivo
que el etileno.
Cuando
dijimos que el carbocatión es la base del mecanismo de la adición
electrofílica, no sólo dimos a entender que es un intermediario, queríamos
indicar que la velocidad de formación
del carbocatión es la que determina el curso de la reacción, como en otras
reacciones carbocatiónicas ya estudiadas.
Ahora
comenzamos a apreciar el recurso poderoso con que contamos para tratar los
problemas que surgen de una amplia gama de reacciones que comprnden
carbocationes. Sabemos que cuanto más estable es el ión, más velozmente se
forma; que su estabilidad depnede de la dispersión de su carga, y que la
dispersión de la carga está determinada por los efectos electrónicos de los
grupos unidos. Ya habíamos descubierto que este mismo enfoque nos permite
tratar con hechos aparentemente tan diferentes como (a) las reactividades
relativas de los sutratos en la sustitución SN1; (b) la relativa facilidad de
deshidratación de los alcoholes; (c) las reactividades relativas de los alquenos hacia la adición de ácidos;
(d) la orientación de la adición de ácidos a los alquenos, y (e) el esquema de
las transposiciones que pueden ocurrir en estas reacciones.
13
Adición de halógenos
El
cloro o el bromo convierten fácilmente a los alquenos en compuestos en compuestos saturados que contienen dos átomos de
halógeno unidos a carbonos adyacentes; generalmente, el yodo no reacciona.
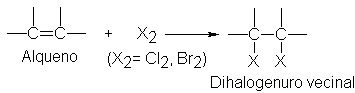
La
reacción se efectúa simplemente mezclando ambos reactivos en un disolvente
inerte como el tetracloruro de carbono. La adición procede velozmente a
temperatura ambiente o inferior, y no necesita exposición a la luz
ultravioleta; de hecho, evitamos deliberadamente temperaturas más altas y
exposición excesiva a la luz, además de
la presencia de un exceso de halógenos, puesto que en tales condiciones la
sustitución podría llegar a constituir una reacción secundaria importante.
 Este proceso, sin duda, es el
mejor método para la preparación de dihalogenuros vecinales. Por ejemplo,
Este proceso, sin duda, es el
mejor método para la preparación de dihalogenuros vecinales. Por ejemplo,
La
adición de bromo es sumamente útil para detectar el doble enlace
carbono-carbono.
Una
solución de bromo en tetracloruro de carbono es roja; el dihalogenuro, igual
que el alqueno, es incoloro. La decoloración rápida de una solución de bromo es
característica de los compuestos que contienen el doble enlace carbono-carbono
14
Mecanismos de la adición de halógenos
Se cree
que la adición de halógenos a alquenos, como la de ácidos próticos, es una
adición electrofílica en dos pasos, y el primero involucra la formación de un
catión, que en la mayoría de los casos no es un carbocatión, sino algo nuevo
para nosotros: un ión halogenonio. Veamos lo que es un ión halogenonio y la
evidencia para su formación.
Utilicemos
la adición del bromo como ejemplo. En
el paso (1), el bromo es transferido deuna molécula de éste al alqueno: no a
uno solo de los carbonos del doble enlace, sino a los dos, formando un ión
bromonio cíclico.
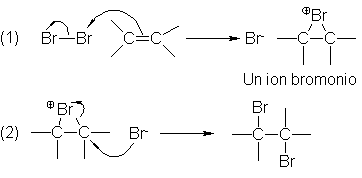
El
paso (1) representa efectivamente una
adición electrofílica. Se transfiere bromo en forma de bromo positivo; es
decir, sin un par de electrones, que quedan en el ión bromuro recién
generado. En el paso (2), este ión
bromuro, o más probablemente otro similar, reacciona con el ión bromonio para
dar el producto: el dibromuro.
Lo que
aquí se propone no es un complejo p, en el que la nube n del
alqueno (base) sujeta a la molécula de bromo (ácido). El bromo está unido con
dos enlaces o uno a cada carbono para establecer un anillo. No obstante, un
complejo n de Br2 molecular y el alqueno puede ser un precursor formado
reversiblemente del ión bromonio.
La
transferencia de un protón de un ácido fuerte a un alqueno, aunque nueva para
nosotros, se ajusta a un marco familiar de reacciones ácido-base. ¿Cómo
interpretaremos la transferencia de un bromo positivo de una molécula del
halógeno? En principio, se trata de una reacción ácido-base, auqnue no en el
sentido de Lowry-Bronsted. Al igual que los alquenos son bases, los halógenos
son ácidos del tipo de Lewis.
Esta
reacción la comprenderemos mejor si modificamos nuestro enfoque. Un ácido es tan sólo en presencia de una base, y
viceversa. De la misma manera, un electrófilo es tan sólo en presencia de un
nucleófilo. Por definición, todo lo que reaccione con un electrófilo tiene que
ser un nucleófilo, y, nuevamente, viceversa. Como químicos orgánicos, tendemos
a hablar de reacciones desde la molécula orgánica, el sustrato. Decimos que
éste sufre una sustitución nucleofílica, el sustrato actúa como electrófilo, y
en la adición electrofílica, como nucleófilo. En el caso de una molécula de
halógeno, la reacción con un alqueno es una sustitución nucleofílica. Actuando como nucleófilo, el alqueno se liga
a uno de los bromos y desplaza al otro en forma de ión bromuro, que es el grupo
saliente y, como ya sabemos, se trata de uno excelente.
¿Cuáles
son los hechos en que se basa el mecanismo?
(a)
el
efeecto de la estructura del alqueno sobre la reactividad;
(b)
el
efecto de nucleófilos agregados sobre los productos obtenidos;
(c)
el
hecho de uqe los halógenos se añaden con estereoselectividad completa, y en el
sentido anti;
(d)
la
observación directa de iones halogenios en condiciones superácidas, y
(e)
la
función de los iones halogenio en los efectos de grupos vecinos.
Los
alquenos, como ya vimos, presenta la misma reactividad a los halógenos que a
los ácidos:los sustituyentes liberadores de electrones activan al alqueno, y
los que atraen electrones, lo desactivan. Este hecho apoya el planteamiento de
que la adición es efectivamente electrófila; que el alqueno actúa como fuente
de electrones y que el halógeno actúa como ácido.
Segundo,
tenemos (b) el efecto de nucleófilos agregados sobre los productos obtenidos.
Si el ión halogenonio capaz de reaccionar con el ión halogenuro es el
intermediario, entonces podemos esperar que reaccione con prácticamente cualquier
ión negativo o molécula básica que le proporcionaremos. El ión bromonio se
forma en la reacción entre etileno y bromo, por ejemplo, y debería ser capaz de
combinarse no sólo con ión bromuro, sino también si estuvieran presentes con iones fluoruro, yoduro y
nitrato, o agua.
Los
hechos concuerdan completamente con esta suposición. Cuando se burbujea etileno
en una solución acuosa de bromo y cloruro de sodio, no sólo se obtiene el
compuesto dibromo, sino también el compuesto bromoclorado y bromoalcohol. El
cloruro de sodio acuoso solo es completamente inerte frente al etileno; cloruro
o el agua sólo pueden reaccionar una vez formado el ión halogenonio por la
acción del bromo. De manera similar, el bromo y el yoduro de sodio acuoso o el
nitrato de sodio convierten al etileno en el compuesto bromoyodado o en el
bromonitrato, además del dibromoderivado y el bromoalcohol, bromo en agua, sin
añadir ningún otro ión, da el derivado dibromado y el bromoalcohol.
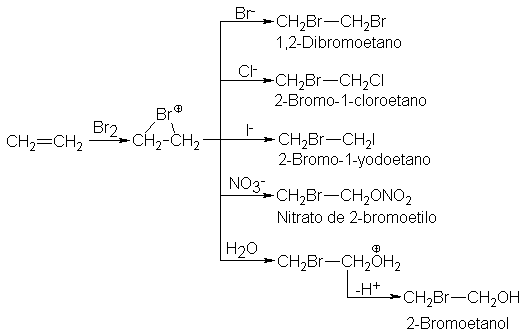
Pues
bien, este trabajo cuidadoso ciertamente demuestra que el etileno reacciona con
bromo para formar algo capaz de reaccionar con los demás nucleóficos pero no es
necesariamente un ión bromonio-. Basados en esta sola prueba, el catión
intermediario podría ser el carbocatión simple abierto BrCH2CH2+. Como veremos
en las secciones 9.5 y 9.6, la estereoquímica de la reacción es la que nos
condujo al concepto de un ión bromonio intermediario, este concepto está
apoyado por la observación de dichos iones.
15
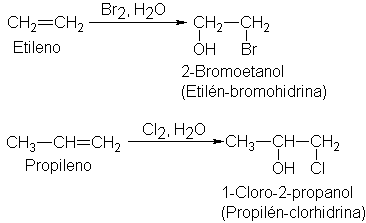
Formación de halohidrinas: adición de los elementos de los ácidos
hipohalogenosos
Vimos
(Sec. 14) que la adición de cloro o bromo, en presencia de agua, puede dar
compuestos que contienen halógeno y un grupo hidroxilo en átomos de carbono adyacentes. Así pues,
estos compuestos son cloro o bromoalcoholes, y suelen llamarse halohidrinas:
cloro o bromohidrinas. En condiciones apropiadas, pueden llegar a ser los
productos principales.
Por
ejemplo,
Existen
pruebas, que no podemos presentar aquí, de que estos compuestos no se forman por adición de los ácidos
hipohalogenosos, HOX, sino por reacción del alqueno con halógeno y agua,
sucesivamente, como se indicó en la sección 8.14.
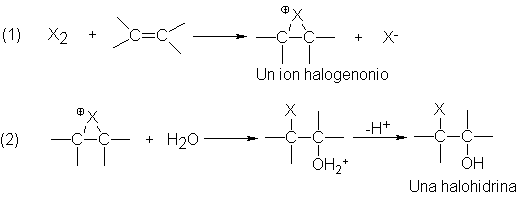
Se
adiciona el halógeno (paso 1) para generar el ión halogenonio, el cual
reacciona, en partes, con agua (paso 2), no con ión bromuro, para dar alcohol
protonado. Cualquiera que sea el mecanismo, el resultado es la adición de los
elementos de un ácido hipohalogenoso (HO- y –X), la reacción se suele citar de
esa manera.
Podemos
apreciar que el propileno da la clorohidrina, en la que el cloro está ligado al
carbono terminal. Este es un comportamiento típico para un alqueno asimétrico;
la orientación sigue la regla de Markovnikov, ya que el halógeno positivo va al
mismo carbono al que iría el hidrógeno de un reactivo prótico.
Pues
bien, esta orientación sería perfectamente comprensible si el intermediario
fuera un carbocatión abierto: la adición inicial de halógeno general el
carbocatión más estable un secundario, en el caso del propileno-. Pero la
estereoquímica indica que en la formación de halohidrinas el intermediario
también es un ión halohenonio cíclico. La orientación depnde de qué carbono
sufre el ataque nucleofílico del agua: el carbono terminal (vía a) o el central
(vía b). El producto obtenido indica que (b) es la vía fuertemente favorecida.
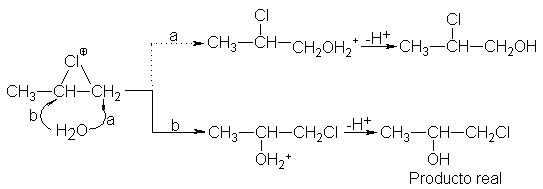
La
estereoquímica de la formación de halohidrinas indica poderosamente que este
ataque ncleofílico es del tipo SN2: la ruptura del enlace carbono-halógeno y la
formación del enlace carbono-oxígeno suceden en un solo paso. Vimos que la
reactividad en SN2 depende típicamente del impedimento estérico . ¿Cómo,
entonces, podemos explicar el ataque preferencial al carbono más impedido del
ión halogenonio?
Ya
habíamos establecido que, en una reacción SN2, el carbono pierde electrones con
el grupo saliente y los gana con el nucleófilo, de modo que el resultado no se
hace apreciablemente positivo o negativo en el estado de transición; los
factores electrónicos carecen de importancia, mientras los estéricos controlan
en gran medida la reactividad.
Sin
embargo, aquí el sustrato es un ión halogenonio. La unión entre carbono y
halógeno es muy débil: en parte, debido a la tensión angular en el anillo de
tre átomos , pero principalmente porque
el halógeno, después de todo, está compartiendo un segundo par de electrones y
soportando una carga positiva. Por esto, el halógeno es un grupo saliente
extremadamente bueno. (Considérese que el grupo saliente no es un ión
halogenuro de por síun buen saliente-. Sino un halógeno neutro, ya ligado a
otro carbono.)
Por
otra parte, el nucleófilo es débil: el agua. Aun cuando en el estado de
transición hay enlaces parcialmente rotos y parcialmente formados, la ruptura
ha avanzado más que la formación de enlaces; el grupo saliente ha quitado
electrones en un grado mucho mayor de los que aporta el nucleófilo, de modo que
el carbono ha adquirido una carga positiva considerable.
La
aglomeración, por otra parte, es relativamente poco importante, debido a que el
grupo saliente y el nucleófilo están lejos. Por tanto, la estabilidad del
estado de transición está determinada principalmente por factoreselectrónicos,
no estéricos. Tal reacción la describimos como de carácter SN1 considerable. El
ataque no ocurre en el carbono menos impedido, sino en el que mejor puede
acomodar una carga positiva.
Así, la
orientación observada es la supuesta si el intermediario fuera un carbocatión
abierto. Veremos que este tipo de orientación se observa corrientemente en
casos como éste, en el que hay un intermediario cíclico de tre átomos con
enlaces débiles al grupo saliente: el
ión mercurinio , por ejemplo, o un
epóxido protonado.
16
Adición de alquenos. Dimerización
En
condiciones apropiadas, el isobutileno es convertido por el ácido sulfúrico o
fosfórico en una mezcla de dos alquenos de fórmula molecular C8H16. La
hidrogenación de cualquiera de estos alquenos produce el mismo alcano
2,2,4-trimetilpentano (Sec. 3.30).Por tanto, ambos alquenos son isómeros que sólo
difieren en la posición del doble enlace. (Problema: Si no fuera así, ¿podrían
ser isómeros geométricos?) Al estudiar estos alquenos por los métodos que se
presentan al final de este capítulo (Sec. 8.23), se encuentra que tienen las
estructuras indicadas:
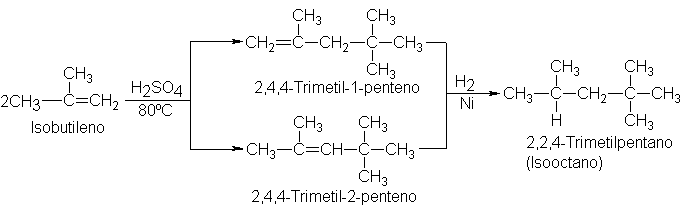
Puesto
que los alquenos producidos tienen exactamente el doble de átomos de carbono e
hidrógeno que el isobutileno original, se conocen como dímeros (di =dos, meros
= parte) del isobutileno, y la reacción se denomina dimerización. Otros alquenos
sufren dimerizaciones similares.
Veamos
si podemos establecer un mecanismo aceptable para esta dimerización. Existe
gran número de octenos isómeros; si nuestro mecanismo nos llevara justo a los
dos que realmente se forman, supondría un apoyo considerable.
Dado
que la reacción es catalizada por ácidos, escribamos como paso (1) la adición
de un ión hidrógeno al isobutileno para formar el carbocatión; el catión
terciario es el ión preferido, por supuesto.
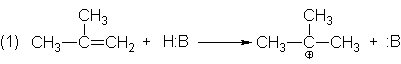
Un
carbocatión sufre reaciones que proporcionan electrones para completar el
octeto del átomo de carbono cargado positivamente. El doble enlace
carbono-carbono es una fuente electrónica excelente, por lo que un carbocatión
bien puede ir hacia él en busca de electrones. En consecuencia, porngamos como
paso (2) la adición del catión t-butilo al isobutileno; la orientación de la
adición nuevamente es tal que resulta el catión terciario más estable. El paso
(2) logra la unión de dos unidades de isobutileno, lo que, desde luego, es
necesario para justificar los productos.
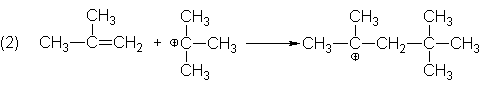
¿Qué es
lo más probable que suceda con el nuevo carbocatión? Podríamos suponer que se
adicione a otra molécula de alqueno, generando un más grande aún; en ciertas
condiciones, esto es lo que sucede. Sin embargo, sabemos que en las condiciones
descritas esta reacción se detiene en compuestos de ocho carbonos, y que éstos
son alquenos.
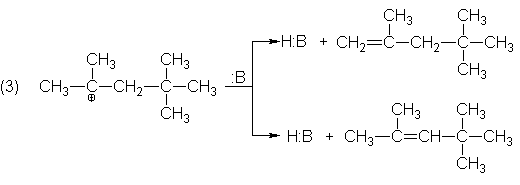 Evidentemente, el carbocatión
sufre una reacción conocida: la pérdida de un ión hidrógeno (paso 3), puesto
que el protón puede perderse en cualqueira de los lados de la carga positiva
debería obtenerse dos productos.
Evidentemente, el carbocatión
sufre una reacción conocida: la pérdida de un ión hidrógeno (paso 3), puesto
que el protón puede perderse en cualqueira de los lados de la carga positiva
debería obtenerse dos productos.
Resulta
que los productos pronoticados para nuestro mecanismo, son precisamente
los que se obtienen. El hecho de que
podamos vaticinarlo, basados tan sólo en las propiedades fundamentales de los
carbocationes tal como las entendemos, evidentemente contituye un apoyo
importante para toda teoría carbocatiónica.
Por lo
que hemos estudiado aquí, podemos agregar una reacción más a la lista. Un
carbocatión puede:
(d)adicionarse
a un alqueno para generar un carbocatión más grande.
Hemos
analizado esta dimerización, no por su gran importancia industrial se sintetiza
isooctano mediante un proceso nuevo de menos costo-, sino por lo que revela
acerca de carbocationes y alquenos. La unión de carbocationes (o especies semejantes) a sistemas de electrones n es
una reacción de un tipo fundamental que se presenta en la química orgánica
común y en la biogénesis en una forma modificada-, la secuencia de reacciones
mediante la cual se forma un compeusto en sistemas vivos, plantas o animales .
17
Adición de alcanos. Alquilación
Veamos
ahora el método industrial que se emplea actualmente para producir grandes
cantidades de 2,2,4-trimetilpentano (isooctano), consumidas en forma de
gasolina de alto octanaje (Sec. 3.30). Al hacer esto aprenderemos más acerca de
las propiedades fundamentales de los carbocationes y algo sorprendente sobre
los alcanos-. Cuando se hacen reaccionar isobutileno e isobutano en presencia de un catalizador ácido, forman directamente
2,2,4-trimetilpentano. Esta reacción es, en efecto, la adición de un alcano a
un alqueno.
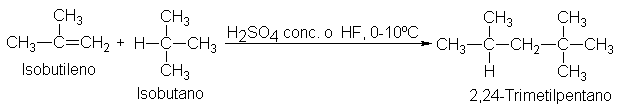
El
mecanismo aceptado comúnmente para esta alquilación se fundamenta en el estudio
de muchas reacciones relacionadas y comprende en el paso (3) una reacción de
carbocationes que no hemos tratado previamente.
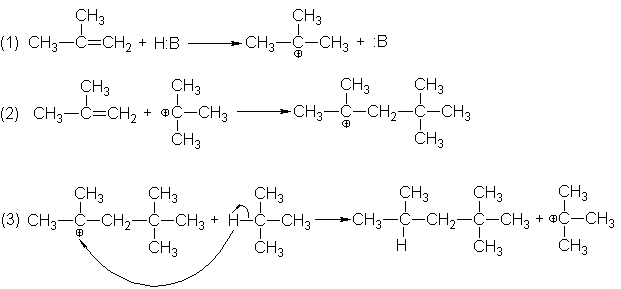
Los dos
primeros pasos son idénticos a los de la reacción de dimerización. En el paso
(3), un carbocatión arranca un átomo de hidrógeno con su par de electrones
(esencialmente un ión hidruro) de una molécula de alcano. Esta separación del
ión hidruro genera un alcano de ocho carbonos y un nuevo carbocatión para
continuar la cadena. Como es de suponer, esta sustracción sucede de modo que
resulta el catión t-butilo en vez del menos estable catión isobutilo (1º).
Este no
es nuestro primer encuentro con la tranferencia de ión hidruro a un carbono
electrónicamente deficiente; vimos prácticamente lo mismo en los
desplazamientos 1,2 que acompañan a las transposiciones de carbocationes (Sec.
5.23). La transferencia allí era intramolecular (dentro de una molécula); aquí
es intermolecular (entre moléculas). Esta reacción demuestra que en un
carbocatión es un ácido muy fuerte. Al mismo tiempo ilustra algo supuesto
anteriormente (Sec. 3.18): que la inercia de los alcanos es muy exagerada. Un
alcano reacciona con facilidad y también en forma heterolítica con un ácido
suficientemente fuerte como reactivo.
Ahora,
actualicemos nuestra lista de reacciones de carbocationes. Un carbocatión puede:
(a)
combinarse
con un ión negativo o una molécula básica;
(b)
transponerse
a un carbocatión más estable;
(c)
eliminar
un protón para formar un alqueno;
(d)
agregarse
a un alqueno para generar un carbocatión más grande;
(e)
quitar
un ión hidruro a un alcano.
Un carbocatión formado según (b) o (d) puede
experimentar luego cualquiera de las reacciones.
Vemos
que todas las reacciones de un carbocatión tienen un fin común: proporcionan un
par de electrones para completar el octeto de un carbono con carga positiva.
18
Adición de radicales libres.
Mecanismo
de la adición de HBr iniciada por peróxidos
En
ausencia de peróxidos, el bromuro de hidrógeno se adiciona a los alquenos de
acuerdo con la regla de Markovnikov; en presencia de aquéllos, se invierte la
dirección de la adición (Sec.6).
Para
explicar este efecto peróxido, Kharash y Mayo propusieron que la adición puede
suceder por dos mecanismos enteramente diferentes: adición según Markovnikov
por el mecanismo iónico que acabamos de estudiar, y adición anti Markovnikov
por un emcanismode radicales libres. Los peróxidos inician la reacción con
radicales libres; en su ausencia o si se agrega un inhibidor (Sec..6, la
adición sigue la vía iónica usual.
La
esencia del emcanismo consiste en que el hidrógeno y el bromo se agregan al
doble enlace homolíticamente, en vez de heterolíticamente; el producto
intermediario es un radical libre, en lugar de un carbocatión. Esta es una
reacción en cadena, al igual que la halogenación de alcanos, pero ahora es una
adición en vez de una sustitución.
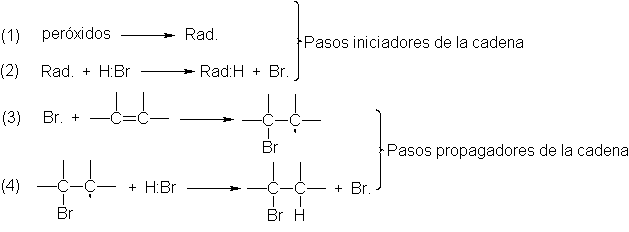
La
descomposición del peróxido (paso 1) para generar radicales libres es una
reacción bien conocida. El radical libre así formado quita hidrógeno al bromuro (paso 2), liberando un a´tomo de
bromo, que se une (paso 3) a uno de los carbonos del doble enlace; al hacer
esto utiliza su electrón impar y uno de los electrones n. El otro carbono queda
con un electrón impar, con lo que el alqueno se convierte en radical libre.
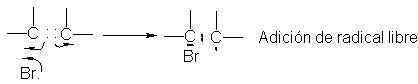
Este
radical libre, al igual que el generado inicialmente del peróxido, le quita
hidrógeno al bromuro de hidrógeno (paso 4). Se completa así la adición,
habiéndose generado un nuevo átomo de bromo para continuar la cadena. Al
igual que en la halogenación de
alcanos, cada cierto tiempo se combina una partícula con otra, o es capturada
por las paredes del reactor, y se interrumpe la cadena.
Este
mecanismo se apoya firmemente en los hechos. El que sólo unas pocas moléculas
de peróxido pueden cambiar la orientación de la adición de muchas moléculas de
bromuro de hidrógeno, es un firme indicio de que se trata de una reacción en
cadena, como lo es el hecho deque unas pocas moléculas de inhibidor pueden
evitar este cambio de orientación. No es de sorprender que estos mismos
compuestos sean inhibidores eficientes de muchas otras reacciones en cadena.
Aunque su modo exacto de accionar se desconoce, parece evidente que interrumpen
la cadena, posiblemente por formación de radicales no reactivos.
No
debemos confundir los efectos de lso peróxidos, que pueden haberse formado por
la acción del oxígeno, con los efectos de este último. Los peróxidos inician
reacciones con radicales libres; el oxígeno, las inhibe (véase Sec. 2.14).
El
mecanismo implica la adición de un átomo de bromo al doble enlace; por tanto,
es apoyado por el hecho de que la adición antiMarcovnikov no sólo es provocada
por la presencia de peróxidos, sino también por irradiación con luz de una
longitud de onda de la que se sabe que disocia el bromuro de hidrógeno
catalizada por luz ha sido estudiada recientemente para varios alquenos por
medio de la espectroscopia RSE (resonancia del espín electrónico), que no sólo puede detectar la presencia de
radicales libres en concentraciones extremadamente bajas, sino que además puede proporcionar
información acerca de sus estructuras .
Se demuestra así la presencia de radicales libres orgánicos en concentraciones
apreciables, lo que concuerda con el mecanismo.
19
Orientación de la adición de radicales libres.
Factores
polares.
¿Cómo
explicamos el hecho de que la adición de radicales libres del bromuro de
hidrógeno sucede con orientación opuesta a la electrofílica? Comparemos ambos
tipos de adición en el propileno.
La
adición electrofílica proporciona bromuro de isopropilo, poeque el catión
isopropilo se genera más velozmente que el catión n-propilo. Esto ya lo hemos
justificado: el catión de isopropilo es el más estable, y los mismos factores
que lo estabilizan, también estabilizan el estado de transición que conduce a
su formación (Sec. 8.12).
La
adición de radicales libres produce el bromuro de n-propilo, porque se genera
más velozmente el radical libre secundario que el primario. Ahora bien, ¿por
qué sucede esto? El estudio de la adición de numerosos radicales libres
diferentes a muchos alquenos distintos incluye tres factores:
(a)
la
estabilidad del radical libre que se está formando;
(b)
factores
polares, y
(c) factores estéricos.
Estudiemos
cada uno de ellos, utilizando como ejemplo la adición de radicales libres del
HBr. Como siempre, cuando nos ocupamos de la velocidad relativa, hay que
considerar el estado de transición para la reacción, y ver cómo puede ser
afectada su estabilidad por cada uno de estos factores.
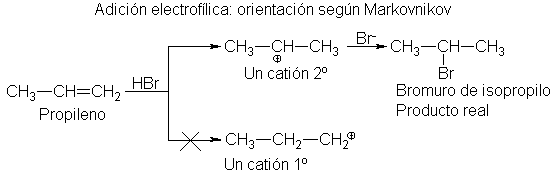

Comencemos
con la estabilidad del radical libre que se forma. Este factor ya es familiar
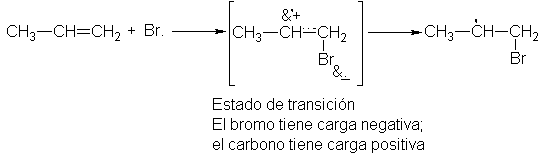
En el estado de transición, el enlace entre el bromo y uno de los carbonos está
parcialmente formado. El enlace n está parcialmente roto, y el otro carbono
tiene ganado, en parte, el electrón impar que portará en el radical libre
intermediario.
El
grupo orgánico posee, en cierto grado, el carácter de radical libre que ha de
llegar a ser.
Los
factores que estabilizan el radical libre también lo hacen con el radical
incipiente en el estado de transición. En nuestro ejemplo, se forma el radical
secundario más velozmente que el primario, porque es más estable. Por tanto,
esta interpretación es la más obvia: la velocidad de reacción depende del
carácter de radical libre del estado de
transición.
Numerosas
observaciones en otros campos de la química de radicales libres han dejado en
claro que las reacciones de radicales libres pueden verse afectadas e incluso
controladas por factores polares (Sec. 36.4). Aun cuando lso radicales libres
son neutros, presentan ciertas tendencias a ganar o perder electrones, por lo
que tienen algo del carácter de reactivos electro o nucleofílicos. Los estados
de transición para sus reacciones pueden ser polares, en los que la parte de
radicales libres adquiere una carga parcial negativa o positiva a expensas del
sustrato.
Ahora
bien, debido a su electronegatividad, sería de esperar que el átomo de bromo
fuese electrofílico. En el estado de transición, el bromo mantiene más de us
cuota de electrones a expensas del alqueno. El estado de ransición es así
porlar, y la aprte de sustrato no sólo tiene carácter de radical libre, sino también carbocatiónico. La
estabilidad del estado de transición, y por tanto la velocidad de reacción,
depende de la capacidad del sustrato para acomodar al electrón impar y la carga
positiva parcial.
El
factor polar favorecerá entonces la orientación que coloque la carga sobre el
carbono que mejor la pueda acomodar. En nuestro ejemplo, la adición de Br. Al
C-1 se ve favorecida, puesto que así se desarolla la carga positiva en el C-2,
en lugar de en el C-1; el carácter de carbocatión secundario es más
estabilizante que el primero.
Por
último, tenemos el factor estérico. La adición de un radical libre al carbono
C-1 terminal está menos impedida que la adición al C-2; el estado de transición
presenta menor aglomeración por lo que es más estable.
En la
reacción particular que estamos estudiando, la adición de radicales libres del
bromuro de hidrógeno, se espera que los tres factores favorezcan la generación
del mismo intermediario y, por consiguiente, den lugar a la misma orientación.
La pregunta es: ¿Cuál es la importancia relativa de cada uno? ¿Cuál es el
factor controlador, si es que hay uno?
La
respeusta es difícil. No cabe duda que existe cada uno de los factores, y de
que puede ser dominante en el sistema adecuado. La adición de radicales libres
a dienos conjugados y estirenos es
evidentemente controlada por la estabilidad del radical en formación. La
orientación de la adición de radicales muy voluminosos, como.CBr3, está determinada
muy probablemente por factores estéricos. La adición de radicales fuertemente
electrófilos, como.CF3, está sometida a efectos polarres marcados en
particular, cuando el alqueno también contiene sustituyentes con tendencias
notorias a la atracción o liberación de electrones.
Sin
embargo, cada uno de estos ejemplos representa un caso extremo: se forma un
radical muy estable; el reactivo es un radical muy voluminoso o muy
electrófilo. ¿Qué nos indica esto en el caso de la adición del átomo de bromo
moderadamente electrofílico y no demasiado grande a un alqueno simple, con formación del único radical secundario
moderadamente estable? Probablemente que los tres factores pueden estar
operando.
La
orientación en la adición electrofílica y en la de radicales libres del bromuro
de hidrógeno está determinada por la formación preferencial de la partícula más
sustituida: un carbocatión o un radical libre. La orientación se invierte,
sencillamente porque se adiciona primero el hidrógeno en la reacción electrofílica;
en la de radicales libres lo hace primero el bromo.
20
Otras adiciones de radicales libres
En los
años posteriores al descubrimiento del efecto peróxido se han encontrado
docenas de reactivos (en su mayoría, por Kharasch), aparte del HBr, que se
adicionan a alquenos en presencia de peróxidos o luz. Para estas reacciones,
también se aceptan mecanismos de radicales libres análogos.
 Así, por ejemplo, para la
adición del tetracloruro de carbono a un alqueno
Así, por ejemplo, para la
adición del tetracloruro de carbono a un alqueno
Otro
ejemplo de adición de radicales libres lo encontraremos en al próxima sección
la polimerización-, reacción que ha sido parte importante de la creación de
esta era de plásticos.
21
Polimerización de alquenos por medio de radicales libres
Cuando
se calienta etileno con oxígeno bajo presión, se obtiene un compuesto de
elevado peso molecular (alrededor de 20 000), que esencialmente es un alcano de
cadena muy larga.
Este
compuesto está formado por muchas unidades etilénicas, y se llama polietileno.
Lo conocemos como material plástico para embalajes.
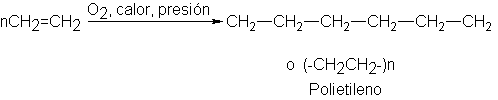
La
formación de polietileno es un ejemplo del proceso llamado polimerización: la
unión de muchas moléculas pequeñas para dar origen a moléculas muy grandes. El
compuesto formado por estas grandes moléculas se denomina polímero (del griego
poli + meros, muchas partes). Los compuestos simples son los que se hacen los
polímeros se conocen como monómeros (mono, uno).
La
polimerización de etilenos sustituidos genera compuestos, cuyas estructuras
presentan cadenas largas del polietileno, con sustituyentes unidos a intervalos
más o menos regulares. Así, por ejmplo, el cloruro de vinilo se convierte en poli(cloruro de vinilo), el cual
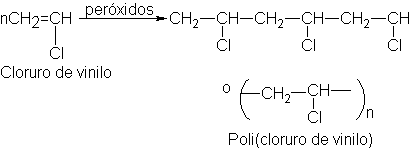
Se
emplea para hacer discos de fonógrafo, mangueras plásticas y plastificado con
ésteres de alto punto de ebullición, impermeables, cortinas para baño,
revestimientos para metales y telas para tapicería.
Muchos
otros grupos (-COOCH3,-CH3,-C6H5, por eejmplo) pueden estar unidos a los
carbonos del doble enlace. Estos etilenos sustituidos se polimerizan con cierta
facilidad, dando origen a plásticos de propiedades físicas y usos diferentes,
pero el proceso de polimerización y la estructura del polímero son básicamente
iguales que para el etileno o el cloruro de vinilo.
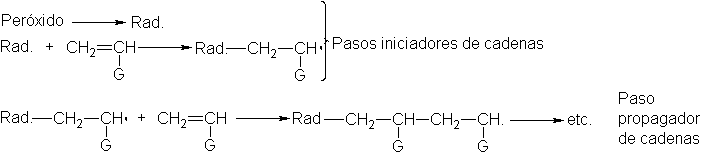 La polimerización debe contar con
la presencia de una pequeña cantidad de un iniciador. Entre los más comunes se
encuentran los peróxidos, que operan rompiéndose para generar un radical libre.
Este radical se une a la molécula del alqueno formado así otro radical libre,
que se agrega a otra molécula de alqueno para generar un radical más grande
que, a su vez, se une a otra molécula de alqueno, y así sucesivamente. La
cadena se termina por pasos, como la unión de dos radicales, que consumen, pero
no generan radicales.
La polimerización debe contar con
la presencia de una pequeña cantidad de un iniciador. Entre los más comunes se
encuentran los peróxidos, que operan rompiéndose para generar un radical libre.
Este radical se une a la molécula del alqueno formado así otro radical libre,
que se agrega a otra molécula de alqueno para generar un radical más grande
que, a su vez, se une a otra molécula de alqueno, y así sucesivamente. La
cadena se termina por pasos, como la unión de dos radicales, que consumen, pero
no generan radicales.
Este
tipo de polimerización, que consume en cada paso una partícula reactiva y
produce otra similar, es un ejemplo de polimerización por reacción en cadena.
En secciones posteriores veremos la polimerización en cadena que no se realiza
por medio de radicales, sino por iones orgánicos, o dentro de la esfera de
coordinación de algún metal complejo de transición.
También
estudiaremos la polimerización por etapas, que implica una serie de reacciones,
cada una de las cuales es, esencialmente, independiente de las otras.
22
Hidroxilación. Formación de 1,2-dioles
Ciertos
agentes oxidantes convierten los alquenos en 1,2-dioles: dihidroxialcoholes que
contienen dos grupos OH en carbonos adyacentes. (También se conocen como
glicoles.)
Su
formación se reduce a la adición de dos grupos hidroxilo al doble enlace.
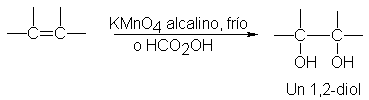
De los
numerosos agentes oxidantes que pueden causar hidroxilación, dos de los
empleados más comúnmente son (a) permanganato de potasio (KmnO4) alcalino y
frío, y (b) peroxi ácidos, como el
ácido peroxifórmico (HCO2OH).
Por ser
el permanganato uno de los agentes oxidantes más importantes en química
orgánica, quizá sea adecuado familiarizarnos ahora con algunas de sus
características generales. Es un oxidante poderoso, por lo que hay que elegir
cuidadosamente las condiciones acidez o alcalinidad, temperatura, cantidad del
reactivo a utilizar para evitar una sobre oxidación; esto es, llevar la
reacción más allá del estado de oxidación deseado. Un problema importante es el
de la solubilidad: poner en contacto el permanganato soluble en agua con el
sustrato, que muchas veces es insoluble en agua. Sin embargo, muchos de los
disolventes que se usan a menudo para juntar reactivos polares y no polares
alcoholes, por ejemplo son oxidados por el empleo de catalizadores de
transferencia de fase Los iones de amonio cuaternarios pueden transportar iones
permanganato de una capa acuosa a una no acuosa (benceno, por ejemplo, o
diclorometano), donde espera el sustrato. Los éteres corona pueden formar
complejos con iones potasio, con lo que el KMNO4 sólido se hace soluble en
benceno; el benceno púrpura resultante es un excelente agente oxidante.
La
hidroxilación con permanganato se lleva a cabo agitando juntos, y a temperatura
ambiente, el alqueno y la solución acuosa de permanganato : esta última
solución es neutra la reacción genera OH-, o mejor, ligeramente alcalina.
Utilizando soluciones de benceno púrpura, se logran, a veces, mejores
rendimientos. Lo importante aquí es la aplicación de condiciones suaves. Se
evita el calor y la adición de ácido, ya que estas condiciones más vigorosas
promueven una oxidación adicional del diol, con escisión del doble enlace
carbono-carbono (Sec. 8.23).
La
hidroxilación con ácido peroxifórmico se lleva a cabo dejando durante unas
horas el alqueno con una mezcla de peróxido de hidrógeno y ácido fórmico,
HCOOH, y luego calentando con agua el producto para hidrolizar ciertos
compuestos intermediarios. Por ejemplo:
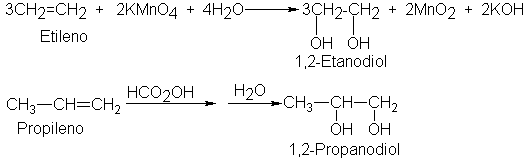
La
hidroxilación de alquenos es el método más importante para la síntesis de
1,2-dioles, con la característica especial de que permite el control
estereoquímico mediante la elección del reactivo (Sec. 19.13).
La
oxidación con permanganato es la base de una prueba analítica útil conocida
como ensayo de Baeyer (Sec.24).
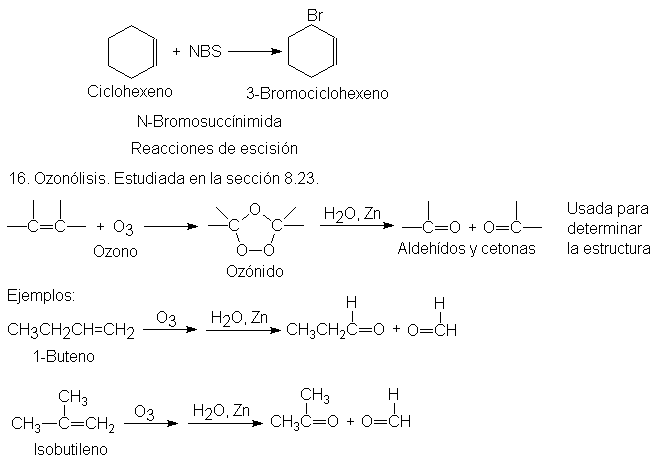
.23
Escisión: determinación de la estructura por degradación. Ozonólisis
Hemos
estudiado las reacciones de adición de los alquenos; en el capítulo 10
trataremos sus reacciones de sustitución. Pero hay un tercer tipo general de
reacción de alquenos, la escisión: una reacción donde se corta completamente el
doble enlace, y la molécula del alqueno se convierte en dos más pequeñas.
El
reactivo clásico para la escisión del doble enlace carbono-carbono es el ozono.
La ozonólisis (ruptura por ozono) se
realiza en dos etapas: la primera es la adición de ozono al doble enlace para
formar un ozónido, y la segunda, la hidrólisis de éste para dar los productos
de la escisión.
Se hace
pasar ozono gaseoso por una solución del alqueno en algún disolvente inerte,
como tetracloruro de carbono; la evaporación del disolvente deja al ozónido en
forma de un aceite viscoso. Este compuesto inestable y explosivo no se purifica
sino que se trata directamente con agua, generalmente en presencia de un agente
reductor.
En los
productos de la escisión se encuentra un oxígeno unido por un doble enlace a
cada uno de los carbonos doblemente enlazador originales.
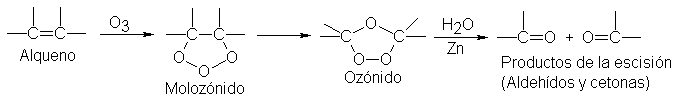
Estos
compuestos que contienen el grupo C = O se llaman aldehídos y cetonas; por el
momento sólo necesitamos saber que son sustancias fácilmente identificables
(Sec. 21.15). La función del agente reductor, que a menudo es polvo de cinc, es
evitar la formación de peróxido de hidrógeno, que puede reaccionar con
aldehídos y cetonas. (Para facilitar el aislamiento, frecuentemente se convierten
los aldehídos, RCHO, en ácidos, RCOOH.)
Conociendo
el número y el ordenamiento de los átomos de carbono de estos aldehídos y
cetonas, podemos reconstruir la estructura del alqueno original. Por ejemplo,
para los tres hexilenos isómeros, tenemos:
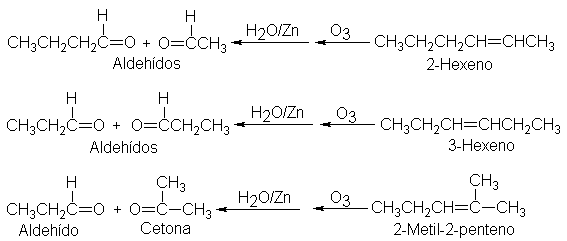
Un
enfoque general para determinar la estructura de un compuesto es la
degradación, o sea, la ruptura de la sustancia desconocida en avrios fragmentos
menores fácilmente identificables. La ozonólisis es un medio de degradación
típico.
Otro
método de degradación, que da esencialmente la misma información, es la
oxidación por peryodato de sodio (NaIO4) en presencia de cantidades catalíticas
de permanganato. El peryodato se utiliza mucho para escindir 1,2-dioles El
permanganato hidroxila el doble enlace (Sec. 8.22) para dar 1,2-diol, con lo
que se reduce al estado de manganato. Lueg, el peryodato (a) escinde el 1,2-dil y (b) reoxida el manganato a
permanganato, por lo que la reacción se sigue desarrollando.
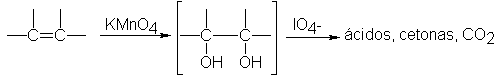
En
lugar de aldehídos, RCHO, se obtienen generalmente ácidos carboxílicos, RCOOH.
Un grupo terminal =CH2 se oxida a CO2. Por ejemplo:
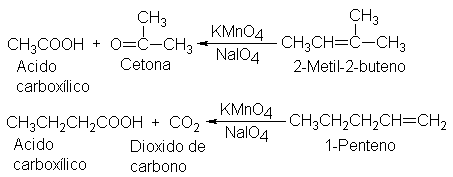
24
Análisis de alquenos
El
grupo funcional de un alqueno es el doble enlace carbono-carbono. Por
consiguiente, para caracterizar un compuesto desconocido como alqueno, debemos
demostrar que sufre las reacciones típicas de ese doble enlace. Puesto que hay
tantas reacciones de ese tipo, podríamos pensar en principio que es una tarea
sencilla; pero consideraremos el problema detenidamente.
En
primer lugar, ¿cuál de todas esas reacciones de alquenos elegimos? ¿Adición de
bromuro de hidrógeno, por ejemplo? ¿Hidrogenación? Imaginemos que estamos
trabajando en un laboratorio con gases, líquidos y sólidos, con matraces, tubos
de ensayo y botellas.
Podríamos
hacer pasar bromuro de hidrógeno seco de un depósito, por un tubo de ensayo que
contiene un líquido desconocido; pero,
¿qué observaríamos? ¿Cómo podríamos decidir si hay reacción o no? Un gas
incoloro burbujea por un líquido incoloro; puede formarse o no un nuevo líquido
incoloro.
Podríamos
intentar la hidrogenación de un compuesto desconocido. Aquí, es posible saber
si hay reacción o no: caída en la presión de hidrógeno indicaría que hubo
reacción.
Esto es
cierto, y la hidrogenación puede ser una herramienta analítica útil, pero hay
que preparar un catalizador y se necesitan aparatos complejos: la operación
completa llevaría horas.
Siempre
que sea posible, seleccionamos para una prueba
de caracterización una reacción que pueda realizarse rápida y
directamente, y que dé origen a un cambio fácilmente observable.
Elegimos
un ensayo que sólo requiera unos minutos, pocos tubos de ensayo, y en el que
haya cambio de color o se produzca el burbujeo de un gas, o se forme o disuelva
un precipitado.
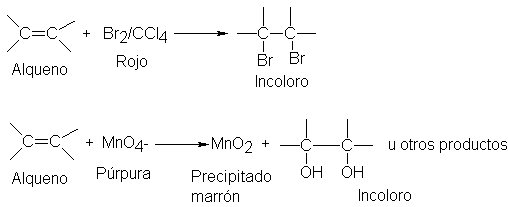 En consecuencia, la experiencia
ha demostrado que la mejor manera de caracterizar un alqueno es por su
propiedad para descolorar una solución de bromo en tetracloruro de carbono
(Sec. 8.13) y una solución diluida, fría y neutra de permanganato (prueba de Baeyer, Sec. 8.22). Ambos ensayos son fáciles de
ejecutar; en uno de ellos desaparece un color rojo, y en el otro, uno púrpura, que es reemplazado por dióxido de
manganeso marrón.
En consecuencia, la experiencia
ha demostrado que la mejor manera de caracterizar un alqueno es por su
propiedad para descolorar una solución de bromo en tetracloruro de carbono
(Sec. 8.13) y una solución diluida, fría y neutra de permanganato (prueba de Baeyer, Sec. 8.22). Ambos ensayos son fáciles de
ejecutar; en uno de ellos desaparece un color rojo, y en el otro, uno púrpura, que es reemplazado por dióxido de
manganeso marrón.
Al
seleccionar los mejores ensayos para la caracterización de alquenos,
consideremos otro problema. Supongamos que agregamos bromo en tetracloruro de
carbono a un compuesto orgánico desconocido y desaparece el color rojo. ¿Qué
indica? Sólo nos dice que nuestra sustancia desconocida reacciona con el bromo;
puede ser un alqueno. Pero no es suficiente saber que un tipo particular de
compuestos se combina con un reactivo determinado: también debemos conocer las
otras clases de sustacnais que tienen el mismo comportamiento. En este caso,
nuestra incógnita puede ser un alquino. (También puede ser uno cualquiera de
una serie de compuestos que sufren sustitución por bromo; sin embargo, en tal
caso se generaría bromuro de hidrógeno, que podría reconocerse por la nube que
forma al echar el aliento sobre el tubo de ensayo.)
De forma
análoga, la decoloración del permanganato no prueba que un compuesto sea un
alqueno, solamente que contiene algún grupo funcional oxidable por el
permanganato. El compuesto puede ser un alqueno, pero también podría ser un
alquino, un aldehído o cualquier otra sustancia fácilmente oxidable. Incluso
puede ser un compuesto contaminado con una impureza oxidable: los alcoholes,
por ejemplo, no se oxidan en las condiciones de este ensayo, pero a menudo
contienen impurezas que sí lo hacen. Generalmente podemos detectar esta
posibilidad asegurándonos que se
decoloren más de una o dos gotas de reactivo.
Por sí
solo, un único ensayo de caracterización raras veces prueba que un compuesto
desconocido sea una clase específica de sustancia.Puede limitar el número de posibilidades,
de modo que la desición final puede tomarse a partir de ensayos adicionales, o
a la inversa, si ya han sido eliminados ciertas posibilidades, una sola prueba
puede permitir que se haga la última elección. De este modo, las pruebas del
bromo y del permanganato serían suficientes para diferenciar un alqueno de un
alcano, un alqueno de un halogenuro de alquilo o un alqueno de un alcohol.
En
consecuencia, los ensayos más utilizados para caracterizar alquenos son: (a)
decoloraciónrápida del bromo en tetracloruro de carbono, sin evolución de HBr,
un ensayo que dan también los alquinos; (b) decoloración de una solución
acuosa, diluida, neutra y fría de permanganato (prueba de Baeyer), un ensayo
que también dan los alquinos y aldehídos. La solubilidadde alquenos en ácido
sulfúrico frío y concentrado es útil, una prueba que también dan muchas otras
sustancias, incluyendo todas las que contienen oxígeno (forman sales de oxonio soluble) y las que se
sulfonan fácilmente (Secs. 15.12 y
28.10). Los alcanos y halogenuros de
alquilo son insolubles en ácido sulfúrico concentrado y frío.(Un ciclopropano
se disuelve fácilmente en ácido sulfúrico concentrado, pero no es oxidado por
permanganato.)
De las
sustancias que ya conocemos, los alcoholes también se disuelven en ácido
sulfúrico, pero pueden distinguirse de los alquenos por dar negativo en las pruebas del bromo en
tetracloruro de carbono y de Baeyer mientras no nos engañen las impurezas. Los
alcoholes primarios y secundarios son oxidados por el anhídrido crómico, CrO3,
en ácido sulfúrico acuoso: en dos segundos, la solución color naranja
transparente se vuelve verde azulada y se opaca.
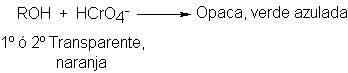
Los
alcoholes terciarios no dan esta prueba, ni tampoco los alcanos.
Una vez
caracterizado como un alqueno, un compuesto desconocido puede identificarse
como un alqueno ya descrito a partir de sus propiedades físicas, incluidos el
espectro infrarrojo y el peso molecular. La comprobación de la estructura de un
compuesto nuevo se consigue mejor por degradación: escisión por ozono
o peryodato/permanganato, seguida de la identificación de los fragmentos formados (Sec. 8.23).
(El
análisis espectroscópico de los qluenos será estudiado en el Cap. 16, en
particular en las Secs. 16.18 y 16.19.)