Alquenos I.
Estructura y preparación
7.1 Hidrocarburos no saturados
7.2 Estructura del etileno. El doble enlace
carbono-carbono
7.4 Hibridación y tamaño orbital
Deshidrogenación: eliminación 1,2 de HX
7.12 Deshidrohalogenación de halogenuros
Deshidrogenación: eliminación 1,2 de HX
7.13 Cinética de la deshidrohalogenación.
7.16 Evidencia para el mecanismo E2. Ausencia
de transposición
7.17 Evidencia para el mecanismo E2. Efectos
isotópicos
7.18 Evidencia para el mecanismo E2.
Ausencia de intercambio de hidrógeno
7.19 Evidencia para el mecanismo E2. El
efecto elemento
7.20 Reacción E2: orientación y reactividad
7.21 Evidencia para el mecanismo E1
7.22 Reaccción E1: orientación
7.23 Eliminación: E2 contra E1
7. 24 Eliminación contra sustitución
7.25 Deshidratación de alcoholes
Eliminación
7.1 Hidrocarburos no saturados
En nuestro estudio de los alcanos mencionados
brevemente otra familia de hidrocarburos, los alquenos, que contienen menos hidrógeno que los alcanos de igual
número de carbonos y que pueden convertirse en éstos por adición de hidrógeno.
Además, se explicó cómo se obtienen los alquenos a partir de los alcanos por
pérdida de hidrógeno en el proceso del cracking.
Puesto que los alquenos tienen,
evidentemente, menos hidrógeno que el máximo posible, se denominan hidrocarburos no saturados. Esta
insaturación puede satisfacer mediante otros reactivos diferentes del
hidrógeno, lo que da origen a sus propiedades químicas características.
7.2 Estructura del etileno. El doble enlace carbono-carbono
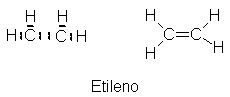
El miembro más sencillo de la familia de los
alquenos es el etileno, C2H4.
En vista de la fácil conversión del etileno en etano, es razonable suponer que
ambos tienen cierta similitud esturctural.
En consecuencia, comenzamos conectando los
átomos de carbono con un enlace covalente y luego unimos a cada uno de ellos
dos átomos de hidrógeno. En esta etapa, cada carbono tiene solamente seis
electrones de valencia, en vez de los ocho requeridos, y a la molécula entera
aún le falta un par para ser neutra. Podemos resolver ambos problemas si damos
por sentado que los carbonos pueden compartir dos pares de electrones y, para
describir esta situación, decimos que están unidos por un doble enlace. El doble enlace carbono-carbono es el rasgo
característico de la estructura de los alquenos.
La mecánica cuántica da una descripción más
detallada del etileno y del doble enlace carbono-carbono. Para formar enlaces
con tres átomos adicionales, el carbono utiliza tres orbitales híbridos
equivalentes sp2, que resultan de mezclar un orbital s y dos
p. Hemos visto (Sec. 1.10) que los orbitales sp2 se encuentran
en l plano del núcleo del carbono y que se dirigen hacia los vértices de un
triángulo equilátero; el ángulo entre cualquier par de orbitales resulta de
120º. Esta disposición trigonal (Fig.
7.1) permite la separación máxima de los orbitales híbridos. Al igual que la
repulsión mutua entre orbitales da cuatro enlaces tetraédricos, también da tres
enlaces trigonales.
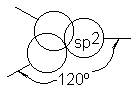
Fig.
7.1 Orbitales atómicos: orbitales híbridos sp2. Los ejes
Se
dirigen a los vértices de un
Tríangulo
equilátero.
Si ordenamos los dos carbonos y cuatro
hidrógenos del etileno para permitir el solapamiento máximo de orbitales,
obtenemos la estructura de la figura 7.2. Cada átomo de carbono se encuentra en
el centro de un triángulo, en cuyos vértices se ubican dos hidrógenos y el otro
carbono. Todos los ángulos de enlace son de 120º. Aunque la distribución en
torno al núcleo de carbono es distinta, individualmente estos enlaces son muy
semejantes a los del etano, siendo cilíndricamente simétricos en torno a una
recta que conecta los núcleos, por lo que se les designa igual: enlace o (enlace sigma).
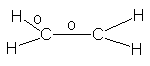
Fig.
7.2 Molécula de etileno: sólo se indican
Los
enlaces o.
Sin embargo, la molécula aún no está
completa. Para formar los tres orbitales sp2 , cada carbono sólo ha
utilizado dos de los tres orbitales p; el restante consta de dos lóbulos
iguales, uno por encima y el otro por debajo del plano de los tres orbitales sp2
(Fig. 7.3), y está ocupado por un solo electrón. Si el orbital p de uno de los
carbonos solapa al del otro, se aparean los electrones y se forman un enlace
adicional.
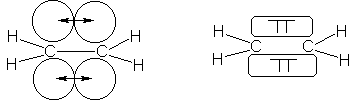
Fig. 7.3 Molécula de etileno: doble enlace
carbono-carbono. El solapa-
Miento de los orbitales p genera un enalce n;
hay una nube n por encima del plano, y otra,
por debajo.
Dado que se genera por solapamiento de
orbitales p y para distinguirla de los
enlaces o, que tienen forma diferente, esta unión se llama enlace n (enlace pi). Consta de dos partes: una nube electrónica
situada por encima del palno de los átomos, y otra, por debajo. Debido al menor
solapamiento, el enlacen es más débil que el o carbono-carbono. En la figura
7.3 podemos apreciar que este solapamiento sólo es factible si los seis átomos
se encuentran en el mismo plano; en consecuencia, la molécula de etileno es plana.
El <<doble enlace>>
carbono-carbono consta, por tanto, de un enlace n débil. La energía total de
enlace (146 kcal) es mayor que la del enlace simple carbono-carbono del etano
(88 kcal). Puesto que los carbonos están conectados más firmemente, la
distancia C-C del etilenoes menor que en el etano, es decir, el doble enlace
carbono-carbono es más corto que el simple.
Se ha estimado que el enlace o del etileno
tiene una energía de unas 95 kcal: es más firme que la del etano, puesto que se
genera por solapamiento de orbitales sp2 (Sec. 7.4). De aquí, es
posible estimar que la energía del enlace n es 51 kcal.
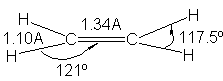
Fig.
7.4 Molécula de etileno: aspecto y di
Mensiones.
Esta estructura mecánico cuántica del etileno
está verificada por pruebas directas:
estudios con difracción electrónica y espectroscópicos indican que la
molécula de etileno es plana (Fig. 7.4), con ángulos de enlace muy cercanos a
120º. La distancia C-C es de 1.34 Å, en comparación con 1.53 Å en el etano
(véase Fig. 7.5).
Además de estas mediciones directas, pronto
veremos que dos aspectos importantes de la química de los alquenos concuerdan
con la descripción mecanicocuántica del doble enlace, en función de la cual
tienen una explicación más simple. Se trata (a) del concepto de rotación impedida y de fenómeno
correspondiente de isomería geométrica
(Sec. 7.6), y (b) del tipo de reactividad química característica del doble
enlace carbono-carbono (Sec. 8.2).
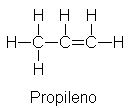
7.3 Propileno
El siguiente miembro de la familia de los
alquenos es el propileno, C3H6.
Dada su gran similitud con el etileno, parece razonable suponer que este
compuesto también tiene un doble enlace carbono-carbono. Partiendo de los dos
átomos de carbono conectados con un enlace doble y enlazando los demás átomos
de acuerdo con la regla de una unión por hidrógeno y cuatro por carbono,
llegamos a la estructura
7.4 Hibridación y tamaño orbital
El doble enlace carbono-carbono de los
alquenos es más corto que el enlace simple correspondiente de los alcanos,
porque cuatro electrones unen más estrechamente que dos. Pero, además, ciertos
enlaces adicionales son apreciablemente más cortos en los alquenos que los
correspondientes en los alcanos; por ejemplo, la distancia C-H en el etileno es
de 1.103 Å, comparada con 1.112 Å, en el etano. Para justificar ésta y otras
diferencias en longitudes de enlaces, debemos considerar diferencias en la
hibridación del carbono.
Los enlaces carbono-hidrógeno del etileno son
simples, como también lo son los del etano, pero aquéllos se originan por
solapamiento de orbitales sp2 del carbono, en vez de sp3,
como en el etano. Comparado con un orbital sp3, uno sp2
tiene menos carácter p y más carácter s. Un orbital p se extiende hasta cierta distancia del núcleo,
mientras que el s está muy cerca de el: a medida que aumenta el carácter s de
un orbital híbrido, disminuye su tamaño efectivo, con lo que también se acorta
la longitud del enlace con otro átomo dado, de dado que un enlace
carbono-hidrógeno sp2-s debería ser más corto que uno sp3-s.
El benceno, cuya molécula es en muchos
sentidos muy distinta que la del etileno (sec. 13.8), también tiene enlaces
carbono-hidrógeno sp2-s de longitud 1.10 Å, casi igual que la del
etileno. El acetileno (Sec. 11.2) contiene carbono con hibridación sp que, en
vista del carácter s aún mayor de sus orbitales debería formar enlaces todavía
más cortos que los del etileno, lo que es correcto, ya que el enlace sp-s mide
solamente 1.079 Å.
Al considerar el tamaño orbital en función de
la hibridación, se llega a la conclusión de que un enlace sp2-sp3
debería ser más corto que un enlace sp3-sp3, lo que es
corroborado por el hecho de que el enlace simple carbono-carbono del propileno
mide 1.501 Å, en comparación con 1.534 Å que mide el enlace correspondiente del
etano. El enlace simple carbono sp-sp3 del metilacetileno (Sec.
10.24) es aún más corto, 1.459 Å. Estas diferencias en las longitudes de los
enlaces simples carbono-carbono son mayores que las correspondientea a los
enlaces carbono-hidrógeno; sin embargo, podría operar otro factor (Sec. 10.23),
aparte de la hibridación particular del carbono.
Una estimación del tamaño orbital en función
de la hibridación nos ayuda a comprender otras propiedades de las moléculas,
además de las longitudes de enlace; por ejemplo, las acideces relativas de
ciertos hidrocarburos (Sec. 11.11) y las basicidades relativas de ciertas
aminas (Sec. 35.11). Es razonable suponer que los enlaces más cortos son más
firmes y, de acuerdo con esto, la tabla 1.2 (Sec. 1.14) indica que la energía
de disociación del enlace C-H para el etileno (108 jcal) es mayor que para el
etano (98 kcal), y la del enlace (simple) C-C es mayor para el propileno (92 kcal) que para el etano (88
kcal). Como se estudiará más adelante (Sec. 10.24), los cambios en la
hibridación pueden ser de importancia más fundamental que la reconocida
generalmente, porque afectan a la estabilidad de las moléculas.
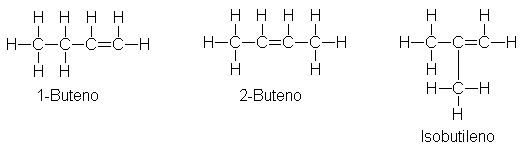
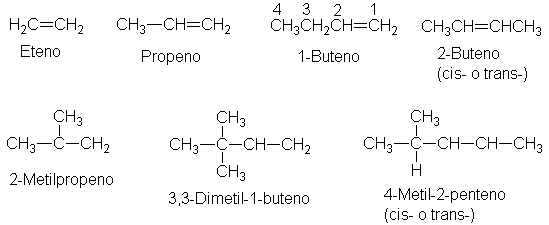
7.5 Los butilenos
Pasando a los butilenos, C4H8, descubriremos que haya
varios ordenamientos posibles. En primer lugar, tenemos un esqueleto de cadena
recta, como el del n-butano, o una estructura ramificada, como la del
isobutano. Luego, y aún restringiéndonos a la cadena recta, observamos dos
disposicionee que difieren en la posición del doble enlace dentro de la cadena.
En consecuencia, tenemos por el momento un total de tres estructuras, que
reciben los nombres 1-buteno, 2-buteno e
isobutileno.
¿Cómo concuerdan los hechos con la predicción
de tres butilenos isómeros? Se ha
demostrado experimentalmente que existen cuatro
alquenos de fórmula C4H8, y no tres, cuyas propiedades
físicas se indican en al tabla 7.1.
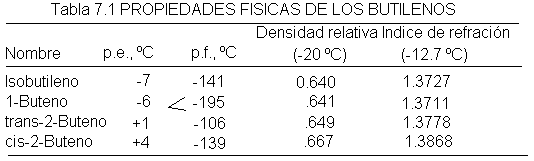
Al hidrogenarlo, el isómero de p.e. –7ºC da
isobutano; evidentemente, este butileno tiene una cadena ramificada, por lo que
su estructura la denominamos isobutileno.
Los otros tres isómeros dan todos el mismo
compuesto por hidrogenación: n-butano; naturalmente, tiene una cadena recta.
Por métodos que estudiaremos más adelante (Sec. 8.23), es posible romper un
alqueno en el doble enlace, y por los fragmentos que se obtienen deducir la
posición del doble enlace en la molécula. Al realizar este proceso, los
productos que resultan del isómero de p.e. –6ºC indican claramente que el doble
enlace está ubicado en un extremo de la cadena, por lo que este butileno tiene
la estructura que llamamos 1-buteno. Al aplicar el mismo procedimiento a los
dos isómeros restantes, ambos dan la misma mezcla de productos, con la que
puede ubicarse el doble enlace en el centro de la cadena.
A juzgar por los productos que se obtienen de
la hidrogenación y de la ruptura, ambos butilenos, el de p.e. + 1 ºC y el de
p.e. +4ºC, tienen la estructura denominada 2-buteno. Sin embargo, las
diferencias en los puntos de ebullición y de fusión y en las demás propiedades
físicas demuestran que se trata de sustancias diferentes, es decir, son
isómeros. ¿En qué pueden diferenciarse sus estructuras?
Para comprender el tipo de isomería que da
origen a dos 2-butenos, debemos examinar con más detenimiento la estructura de
los alquenos y la naturaleza del doble enlace carbono-carbono. El etileno es
una molécula plana que resulta así por la disposición geométrica de los
orbitales enlazantes y, en particular, por el solapamiento que origina el
orbital n. Por las mismas razones, la porción que comprende los dos carbonos
con doble enlace y los cuatro átomos unidos a ellos debe ser plana en todo
alqueno.
Si examinamos con mayor detalle la estructura
del 2-buteno, especialmente si usamos modelos moleculares, observaremos que hay
dos formas bien diferentes, I y II, de ordenar los átomos (aparte de las
infinitas posibilidades que surgen del giro en torno a enlaces simples). En una
de las estructuras, los grupos metilo se encuentran en el mismo lado de la
molécula (I), y en la otra, en lados opuestos (II).
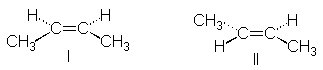
Surge ahora una pregunta: ¿Podemos aislar dos
2-butenos isómeros que correspondan a estas dos estructuras diferentes, o se
interconvierten demasiado fácilmente: como las conformaciones del n-butano, por
ejemplo (Sec. 3.5)?
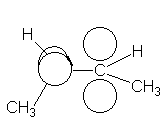
Fig.
7.6 Rotación impedida en torno al doble-
Enlace
carbono-carbono. El giro evita el
Solapamiento
de orbitales p, rompiendo el enlace n.
La
conversión de I en II implica la rotación en torno al doble enlace
carbono-carbono, y la posibilidad de aislar isómeros depende de la energía
requerida para esta rotación. Hemos visto que la formación del enlace n
involucra el solapamiento de orbitales p, ubicados por encima y por debajo del
plano de los orbitales o. Para pasar de uno de estos 2-butenos al otro, debe
torcerse la molécula para que ya no se
solapen los orbitales p, es decir, debe romperse el enlace n (véase Fig. 7.6).La
ruptura del enalce n requiere unas 70
kcal; a temperatura ambiente, sólo una proporción insignificante de colisiones
tiene esta energía cuenca, debido a esta barrera energética de 70 kcal, la rotación en torno al doble enlace carbono-carbono está impedida. Como resultado
de esta rotación impedida, pueden aislarse dos 2-butenos isómeros, que son,
claro está, los butilenos de p.e. +1 ºC y p.e. +4 ºC.
7.6 Isomería geométrica
Puesto que los 2-butenos isómeros sólo difieren en la orientación espacial
de sus átomos (pero son semejantes en cuanto a qué átomos se unen a cuáles
otros), pertenecen a la clase general de isómeros que denominamos estereoisómeros
(Sec. 4.1). Sin embargo, no son imágenes especulares entre sí, por lo que no
son enantiómeros. Como ya sabemos, los
estereoisómeros que no son imágenes especulares mutuas se llaman diastereómeros.
El tipo específico de diastereómeros que
deben su existencia a la rotación impedida en torno a dobles enlaces se conoce
como isómeros geométricos. Por
consiguiente, los 2-butenos isómeros son diastereómeros, más específicamente,
isómeros geométricos.
Recordamos que la disposición característica
de los átomos de un estereoisómero es su
configuración; las de los 2-butenos isómeros son las estructuras I y II,
cuyos nombres se diferencian por los prefijos cis- (del latín, <<a este lado>>) y trans- (del latín, <<al otro
lado>>), que indican que los grupos metilo se encuentran al mismo lado o
en lados opuestos de la molécula. Por un método que veremos un poco más
adelante (Sec. 7.9), al isómero de p.e.
+4 ºC se le ha asignado la configuración cis, y al de p.e. +1 ºC, la
configuración trans.
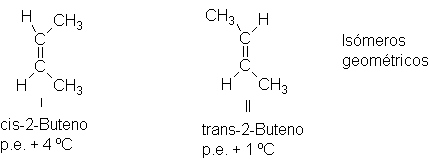
En la figura 7.7 se observan modelos a escala
de los 2-butenos isómeros. En el isómero trans,
los grupos metilo están muy separados, mientras que en el cis se encuentran
cerca, lo suficiente para provocar una aglomeración. Por tanto, podemos suponer
que el isómero cis es menos estable que el trans,
y veremos que es así (Sec. 8.4).
La rotación se halla impedida en torno a cualquier doble enlace carbono-carbono,
pero solamente origina isomería geométrica si se cumplen ciertas relaciones
entre los grupos unidos a los carbonos con doble enlace. Podemos reconocer esta
isomería dibujando las posibles estructuras (o, mejor aún, construyéndolas con
modelos moleculares) y luego
comparándolas, para ver si efectivamente son isómeras o idénticas. De este
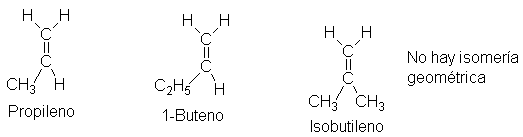
modo, llegamos a la conclusión de que el propileno, el 1-buteno y el
isobutileno no deberían presentar isomería, lo que concuerda con la realidad.
Es evidenteque muchos alquenos superiores exhiben isomería geométrica.
Si consideraremos compuestos distintos de los
hidrocarburos, resulta que el 1,1-dicloro-eteno y el 1,1-dibromoeteno no
deberían presentar isomería, mientras que los 1,2-dicloro-y
los
1,2-dibromoetenos, sí. En todos los casos estos pronósticos resultaron
correctos, habiéndose aislado isómeros con las propiedades físicas siguientes:
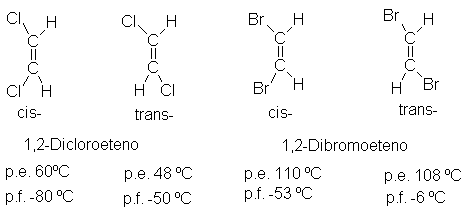
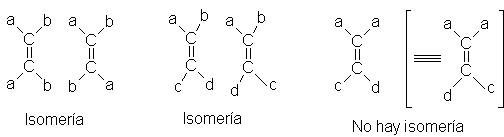
Del examen de estas estructuras, podemos
concluir que la isomería geométrica no puede existir si cualquiera de los
carbonos lleva dos grupos idénticos. A continuación se muestrasn algunas
combinaciones posibles.
El fenómeno de la isomería geométrica es
general y puede encontrarse en cualquier clase de compuestos con dobles enlaces
carbono-carbono (e incluso con dobles enlaces de otros tipos).
Los prefijos cis y trans son adecuados para etilenos disustituidos y
algunos trisustituidos; pero, ¿cómo
hemos de especificar configuraciones como las siguientes? ¿Qué grupos son
nuestros puntos de referencia? Observando por separado cada uno de los carbonos
del doble enlace, ordenamos sus dos átomos o grupos de acuerdo con la secuencia
de Cahn-Ingold Prelog (Sec. 4.16).A continuación, elegimos el grupo prioritario
de cada uno de los carbonos y determinaremos si ambos se encuentran al mismo
lado de la molécula o si están en lados opuestos. Para dar por sentado que
estamos empleando este método de especificación,
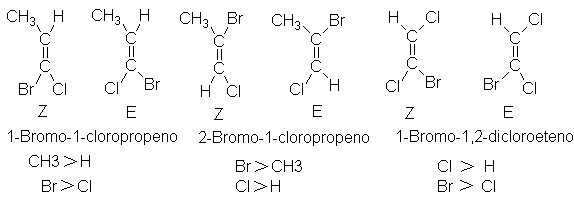
usamos la letra Z para indicar al
mismo lado y la letra E para indicar lados
opuestos. (Del alemán, zusamente,
junto, y entgegen, opuesto.) La letra
apropiada pasa entonces a ser parte del nombre de ese alqueno:
(Z)-1-bromo-1-cloropropeno, por ejemplo.
En consecuencia, un par de isómeros
geométricos son diastereómeros. ¿Donde encajan en el otro sistema de
clasificación, el basado en cómo se interconvierten los estereoisómeros (Sec.
4.20)? Como hemos visto, hay:
(a) isómeros configuracionales, que se interconvierten por inversión (se dan vuelta) en un centro
quiral: y
(b) (b) isómeros conformacionales,
interconvertibles por rotación en torno a enlaces simples.
Ahora bien, agregamos:
(c) isómeros geométricos, interconvertibles en principio por rotación en torno a un doble
enlace.
La operación requerida una rotación es la
misma para la interconversión de isómeros geométricos y conformacionales. Desde
el punto de vista eminentemente práctico de la aislabilidad, los isómeros geométricos son más afines con los
configuracionales: la interconversión requiere una ruptura de enlaces un enlace
n en el caso de los isómeros geométricos y, en consecuencia, es siempre un
proceso difícil. Los isómeros conformacionales se interconvierten por el
proceso (generalmente) sencillo de la rotación en torno a enlaces simples.
Por conveniencia, hemos formulado la
siguiente <<regla básica>> para los planteamientos y problemas de
este texto: a menos que se indique expresamente de otra manera, los términos
<<estereoisómeros>>, <<enantiómeros>> y
<<diastereómeros>> solamente se referirán a isómeros
configuracionales y geométricos, excluyendo los conformacionales. Estos
últimos se denominarán i<<sómeros conformacionales>>,
<<confórmeros>>, <<enantiómeros conformacionales>> y
<<diastereómeros conformacionales>>.
En lo que respecta a las propiedades químicas
y físicas, los isómeros geométricos guardan la misma
relación entre sí que los demás diastereómeros estudiados (Sec. 4.17).
Tienen los mismos grupos funcionales, por lo
que exhiben propiedades químicas similares; no
son idénticas, sin embargo, puesto que sus estructuras ni son idénticas ni
son imágenes especulares; se combinan con los mismos reactivos, pero con
velocidad diferente. (Como veremos en el Cap. 9, en ciertas condiciones
esepcialmente en sistemas biológicos el comportamiento químico de los isómeros
geométricos puede ser muy diferente.)
Tal como ilustran los ejemplos anteriores.
Los isómeros geométricos tienen propiedades físicas diferentes: distintos
puntos de ebullición y de fusión, índices de refracción, solubilidades,
densidades, etc. Basándonos en estas propiedades físicas diferentes, se pueden
distinguirse unos de otros e identificarse, una vea establecida la
configuración de cada uno. AL menos en principio, también son separables (véase
Sec. 4.17).
Cuando estudiemos las propiedades físicas de
los alquenos (Sec. 7.9), veremos uno de los medios para determinar si una
sustancia particular es el isómero cis o
trans; es decir, uno de los métodos para asignar configuraciones.
7.7 Alquenos superiores
Pudimos apreciar que los butilenos tienen un
carbono y dos hidrógenos más que el propileno, que, a su vez, tiene un carbono
y dos hidrógenos más que el etileno; por tanto, los alquenos forman otra serie
homóloga con el mismo incremento que los alcanos: CH2. La fórmula
general para esta familia es CnH2n.
A medida que avanzamos en la serie de los
alquenos, el número de estructuras isómeras para cada miembro aumenta aún más
rápidamente que en el caso de la serie de los alcanos; además de las
variaciones en los esqueletos carbonados, hay variaciones en la posición del
doble enlace dentro de ellos, como también hay posibilidad de isomería
geométrica.
7.8 Nombres de los alquenos
Raras veces se usan nombres comunes, salvo
tres alquenos sencillos: etileno,
propileno e isobutileno. Sin embargo, los
diversos alquenos de un determinado número de carbonos suelen designarse
colectivamente como pentilenos (amilenos),
hexilenos, heptilenos, etcétera. A
veces, se encuentran alquenos denominados como derivados del etileno, por
ejemplo, tetrametiletileno por (CH3)2C=C(CH3)2. La mayoría
se designa por medio del sistema IUPAC, cuyas reglas son:
1. Selecciones como estructura de referencia la cadena continua más larga
que contenga el doble enlace; luego,
considérese el compuesto como derivado de aquella estructura por reemplazo de
hidrógenos por diversos grupos alquilo. La estructuraa matriz se
2. conoce como eteno, propeno,
buteno, penteno, y así sucesivamente, según sea su número de átomos de
carbono; cada nombre se obtiene cambiando la terminación –ano del alcano
correspondiente a –eno:
3. Indíquese la ubicación del doble enlace en la cadena matriz por medio
de un número. Aunque el doble enlace abarca dos carbonos, fijese su posición
con el número correspondiente al primer
carbono del doble enlace; la numeración de la cadena comienza desde el extremo
más cercano al doble enlace: es decir, 1-buteno
y 2-buteno.
3.Por medio de números, indíquense las
posiciones de los grupos alquilo unidos a la cadena principal.
Cuando ha de especificarse un isómero
geométrico, se añade un prefijo: cis- o
trans-, o (Z)- o (E)-.
Un alqueno que contiene halógeno generalmente
recibe el nombre de haloalqueno,
esto es, un alqueno que contiene halógeno como cadena. Hay dos grupos no
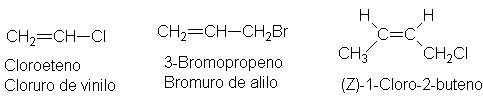
saturados tan comunes que se les da un nombre especial: vinilo, CH2=CH-, y alilo,
CH2=CH-CH-CH2-.
7.9 Propiedades físicas
Como clase, los alquenos poseen propiedades
físicas esencialmente iguales que las de los alcanos. Son insolubles en agua,
pero bastante solubles en líquidos no polares, como benceno, éter, clororformo
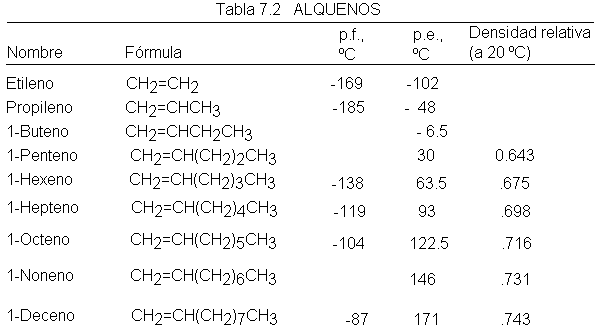
o ligroína, y son menos densos que el agua. De la tabla 7.2 se desprende que el
punto de ebullición aumenta con el número creciente de carbonos; como en el
caso de los alcanos, el aumento del punto de ebullición es de 20 a 30 grados
por cada carbono adicional, excepto para los homólogos muy pequeños. Las
ramificaciones bajan el punto de ebullición. Una comparación de la tabla 7.2
con la 3.3 (Sec. 3.12) demuestra que el punto de ebullición de un alqueno es
muy parecido al del alcano con un esqueleto carbonado correspondiente.
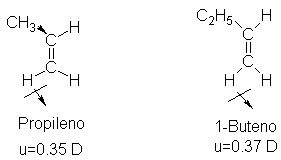
Al igual que los alcanos, los alquenos son, a
lo sumo, sólo débilmente polares. Puesto que los electrones n muy sueltos del
doble enlace se desplazan con facilidad, sus momentos dipolares son mayores que
los de los alcanos; sin embargo, son pequeños: por ejemplo, compararemos los
momentos dispolares del propileno y del 1-buteno, señalados más adelante, con
el del cloruro de metilo, 1.83 D. La unión del grupo alquilo al carbono del
doble enlace tiene una polaridad pequeña, cuya dirección se supone que es la
indicada, es decir, con el
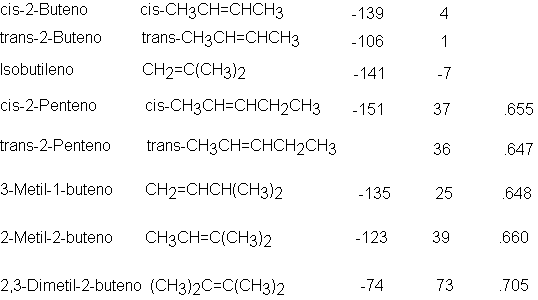
Alquilo
desprendiendo electrones hacia el doble enlace. Puesto que esta
polaridad no es anulada por una equivalente en dirección contraria, confiere a
la molécula un momento dipolar neto.
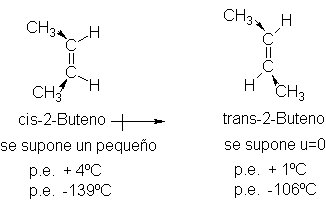
El cis-2-buteno,
con dos grupos metilo a un lado de la molécula y dos hidrógenos al otro, debe
exhibir un pequeño momento dipolar; por el contrario, en el trans-2-buteno, con un metilo y un hidrógeno a cada lado de su
molécula, los momentos de enlace deben anularse. Aunque sus momentos dipolares
no han sido medidos directamente, se refleja una pequeña plaridad en el punto
de ebullición más alto del isómero cis.
Igual relación existe para muchos pares de
isómeros geométricos. Por su mayor polaridad, generalmente el isómero cis es el de punto de ebullición más
elevado de un par, y por su menor simetría, se acomoda más imperfectamente en
el retículo cristalino, por lo que generalmente tiene el punto de fusión menor.
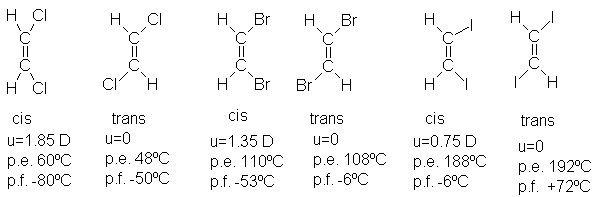
La diferencia en plaridad, y en consecuencia
en puntos de fusión y ebullición, son mayores para los alquenos que contienen
elementos cuyas electronegatividades son muy distintas de la del carbono. Por
ejemplo:
La relación entre configuración y puntos de
ebullición o fusión es sólo una regla empírica para la que existen muchas
excepciones (por ejemplo, los puntos de ebullición de los diyodoetenos). Por
otra parte, la maldición de los momentos dipolares frecuentemente permite
determinar claramente si un isómero dado es cis
o trans.
7.10 Fuente industrial
El petróleo y el gas natural proporcionan los
alcanos que son la principal fuente primaria de productos orgánicos: sustancias
en torno a las que se ha desarrollado una industria vastísima, y que utilizamos
en laboratorio. Ahora bien, los alcanos propiamente dichos no se prestan a la
conversión directa en diversos compuestos: son relativamente inertes, y las
reacciones que pueden sufrir se realizan
más o menos indiscriminadamente sobre la molécula para generar mezclas
complejas.
Sin embargo, de los alcanos se obtienen,
mediante el cracking en sus diversas
formas (Sec. 3.31), ciertas sustancias más reactivas: los hidrocarburos aromáticos benceno, tolueno y los xilenos (Cap. 15);
y los alquenos menores etileno,
propileno y los butilenos. En último término, la mayoría de los productos
aromáticos y alifáticos se obtiene de estos pocos compuestos más metano. El
etileno, por ejemplo, es el compuesto orgánico de mayor consumo en la industria
química y se sitúa en quinto lugar entre todos, siendo superado solamente por
el ácido sulfurico, la cal, el amoniaco y el oxígeno.
Al contrario de los alcanos, los alquenos son
muy reactivos, debido a su grupo funcional: el doble enlace carbono-carbono.
(En realidad, los alcanos no poseen grupo funcional o bien, si lo tienen, es H,
que se encuentra por todos lados en la molécula-.)
Los alquenos so sólo sufren una amplia gama
de reacciones, sino que, además, éstas ocurren
en lugares bien definidos de la molécula: en el propio doble enlace o en
ciertas posiciones que poseen una relación específica con respecto a áquel. Las
condiciones en las que los alquenos reacionan a escala industrial pueden, por
razones prácticas y económicas, diferir ampliamentede las que se utilizan en
laboratorio; sin embargo, en el análisis final, las reacciones que de hecho se
realizan son las mismas que estudiaremos en el capítulo 8.
En el grado en que la biomasa renovable algún
día reemplace a la masa fósil, no renovable, como fuente primaria de productos
químicos orgánicos, sin duda los alquenos continuarán teniendo un papel
primordial. El etileno, por ejemplo, se forma con facilidad por deshidratación
del etanol, el cual se produce por la fermentación de carbohidratos.
7.11 Preparación
Los alquenos no superiores a cuatro carbonos
pueden obtenerse puros en la industria petrolera. Muestra de alquenos puros más
complejos se deben preparar por métodos como los que describiremos más
adelante.
La introducción de un doble enlace
carbono-carbono en una molécula que sólo tiene enlaces simples supone
necesariamente la eliminación de
átomos o grupos de dos carbonos adyacentes:
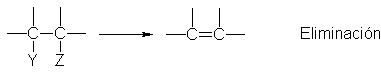
En el proceso del cracking citado, por ejemplo, los dos átomos eliminados son
hidrógeno:
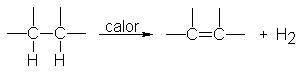
Las reacciones de eliminación que se
describen a continuación pueden utilizarse no sólo para obtener alquenos
simples, sino también lo que es mucho más importante para suministrar los
métodos generales más adecuados de introducir dobles enlaces carbono-carbono en
todo tipo de molécula.
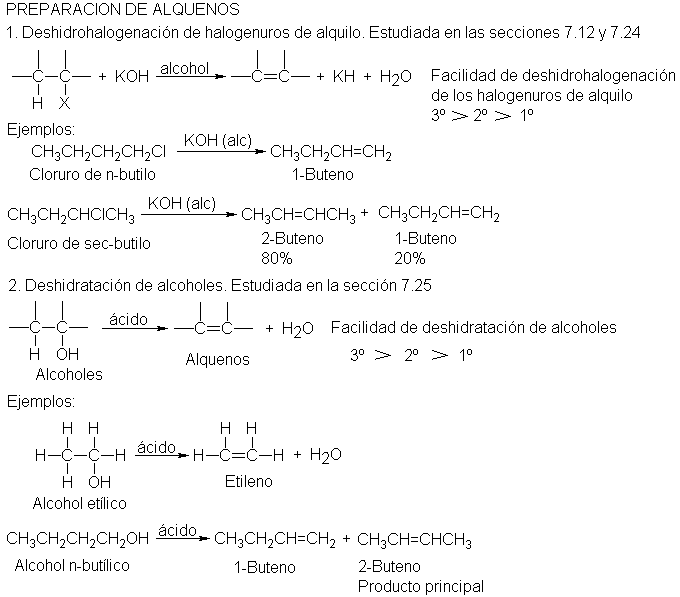
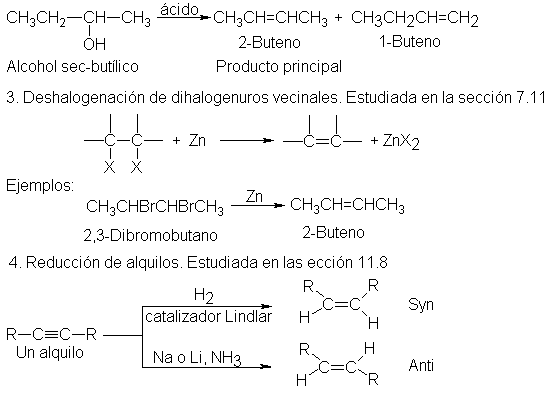
Los más importantes de estos métodos de
preparación son su aplicabilidad más general la deshidrohalogenación de los halogenuros de alquilo, promovida por
una base,
Deshidrogenación: eliminación 1,2 de HX
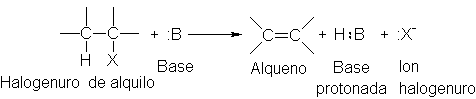
Y la deshidrogenación
de alcoholes, catalizada por un ácido.
Deshidratación: eliminación 1,2 de H2O
Lógicamente, los sulfonatos de alquilo sufren
una eliminación, promovida por bases, muy similar a la deshidrohalogenación: lo
que hay que decir sobre la deshidrogenación también es aplicable a esta
reacción.
Ya vimos (Sec. 5.7) que los halogenuros y
sulfonatos de alquilo se preparan casi siempre a partir de los alcoholes
correspondientes, por lo que todos estos métodos involucran, en esencia, la
preparación a partir de alcoholes; no obstante, la eliminación promovida por
bases causa, generalmente, menos complicaciones, por lo que es a menudo el
método preferido, a pasar del paso adicional en la secuencia.

Tanto la deshidrohalogenación como la
deshidratación tienen el inconveniente de que pueden, donde la estructura lo
permita, eliminar hidrógeno del carbono en cualquiera de los lados del carbono
que tiene el X o el -OH, lo que frecuentemente produce isómeros. Para asegurar
que el doble enlace aparece sólo entre un par determinado de átomos de carbono,
necesitamos un sustrato del que únicamente se pueda eliminar un par dado de
sustituyentes ninguno de ellos el omnipresente hidrógeno-, como en la reacción de Wittig,, por ejemplo (Sec.
25.10).
La deshalogenación de dihalogenuros vecinos
está muy limitada por el hecho de que esos dihalogenuros se preparan
generalmente a a partir de alquenos. Sin embargo, puede ser útil transformar un
alqueno en un dihalogenuro mientras se realiza algún cambio en otra parte de la
molécula, y después regenerar el alqueno al tratar el dihalogenuro con Zn; este
procedimiento se conoce como protección
del doble enlace.
Al igual que es posible generar un doble
enlace carbono-carbono de uno simple por eliminación, también se puede generar
por adición a partir de un enlace triple carbono-carbono.
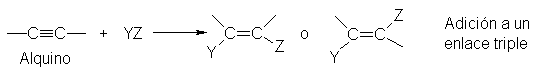
Dado que tal adición a menudo puede
controlarse para producir un alqueno cis o trans, alqueno según se desee, los
compuestos con enlace triple (los alquinos,
Cap. 11) son intermediarios importantes en la síntesis de alquenos cis o trans estereoquímicamente puros.
Los intermediarios clave en las síntesis
anteriores son alcoholes y alquinos. Como veremos, ambos tipos de compuestos
son, a su vez, fácilmente accesibles partiendo de sustancias más simples.
Combinando la química de los alquenos con la de los alcoholes y alquinos,
obtendremos alquenos en una amplia variedad de formas y tamaños.
7.12 Deshidrohalogenación de halogenuros
de alquilo: eliminación 1,2
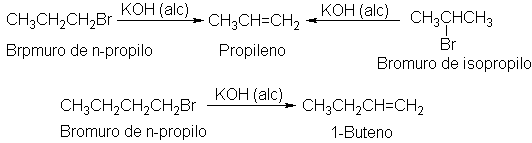
Cuando se trata bromuro de isopropilo con una
solución alcohólica concentrada y caliente de una base fuerte, como el
hidróxido de potasio, se obtiene propileno, bromuro de potasio y agua.
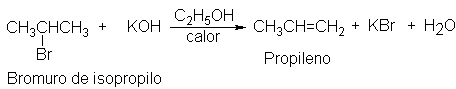
Este es un ejemplo de deshidrohalogenación: una
eliminación 1,2 de los elementos de
un halogenuro de hidrógeno. La deshidrohalogenación implica la pérdida
eliminación del átomo de halógeno y de uno de hidrógeno de un carbono adyacente
al que pierde el halógeno. El reactivo requerido es una base, cuya función es extraer un hidrógeno en froma de protón. (La
base :B puede ser neutra o tener una carga negativa, por ejemplo, H2O
o OH-. Entonces, el ácido conjugado tendrá un acarga positiva o será
neutro, por ejemplo, H3O+ o H2>>.)
Deshidrogenación: eliminación 1,2 de HX
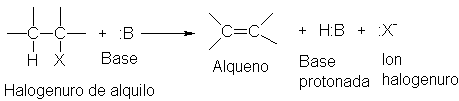
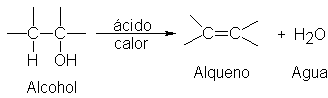
¿Cómo genera tal eliminación un doble enlace?
Con independencia del mecanismo exacto, los productos de la reacción indican
que debe suceder lo siguiente: el halógeno abandona la molécula como ion
halogenuro, por lo que debe llevar consigo su par de electrones. El hidrógeno
es extraído por la base como un protón, de modo que debe abandonar su parte de
electrones; éste es el par que queda disponible para fromar el segundo enlace
(el enlace n) entre los átomos de carbono.
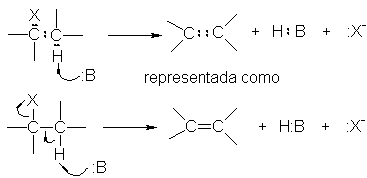
Donde las flechas indican la dirección del
desplazamiento de electrones
A estas alturas, lo que hemos ilustrado
intenta ser sólo una especie de conteo de electrones, para mostrar los cambios
en los enlaces el movimiento de los electrones que deben ocurrir. De hecho, representa
un mecanismo específico de hecho, el más común de ellos en el que el proceso
completo se desarrolla en una sola etapa, donde suceden simultáneamente las
rupturas y formaciones de enlaces. Veremos, sin embargo, que hay otros
mecanismos que debemos considerar: mecanismos en los que ocurren los mismos
cambios, pero con diferente programación en el tiempo. Como en la sustitución nucleofílica y en muchas de las
otras reacciones que estudiaremos, el tiempo es a menudo el punto crucial en
que difieren los distintos mecanismos, y para establecerlo se reúne gran parte
de la evidencia experimental.
A esto le hemos dado el nombre de eliminación
1,2: para que se establezca el doble enlace, el hidrógeno debe proceder de un
carbono adyacente al que tiene el halógeno. El
carbono que tiene el halógeno se suele denominar carbono a (carbono alfa).
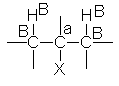
Todo carbono unido al a es un carbono ß (beta), y sus hidrógenos también son
hidrógenos ß. Por tanto, la eliminación
implica la pérdida de un hidrógeno B.
En algunos casos, la eliminación genera un
solo alqueno,
Y en otros, resulta una mezcla.
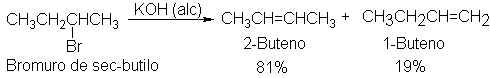
Para predecir qué productos se formaránm en
una reacción determinada, basta con examinar elsustrato. Podemos esperar la
obtención de un alqueno correspondiente a la pérdida de cualquiera de los hidrogeno ß-pero
ningún otro alqueno-. El bromuro de n-butilo, por ejemplo, sólo puede
perder hidrógeno del C-2,
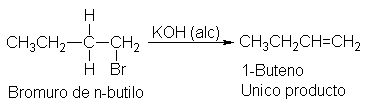
De modo que da 1-buteno, solamente. El
bromuro de sec-butilo, por otra parte, puede perder hidrógeno del C-1,
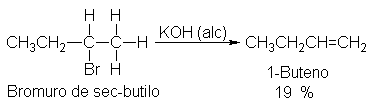
O del C-3,
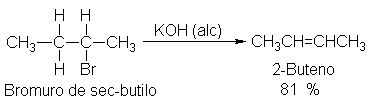
Por lo que da 1- y 2-buteno. En donde pueden
formarse ambos alquenos, el producto principal es el 2-buteno: este hecho se
ajusta a un esquema general para la deshidrohalogenación, que trataremos más
adelante (Sec. 7.20).
(Lo que acabamos de ver supone que no ha
habido transposición, una suposición justificada por la deshidrohalogenación
llevada a cabo en condiciones usuales: en soluciones alcohólicas concentradas
de base fuerte. Aprenderemos a reconocer situaciones en las que las
transposiciones son probables y a prededcir también los productos de
eliminación en aquellos casos.)
Con el estudio fr la deshidrohalogenación
aprenderemos bastante acerca de toda la clase de reacciones a la cual
pertenece, y de la cual es típica: la eliminación
1,2.
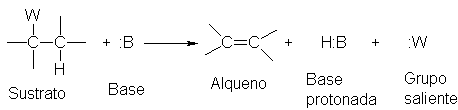 Eliminación 1,2
Eliminación 1,2
Estas reacciones de eliminación se
caracterizan por lo siguiente:
(a) El sustrato contiene un grupo
saliente, un átomo o grupo que abandona la molécula, llevándose un par de
electrones.
(b) En una posición beta con
respecto al grupo saliente, el sustrato contiene un átomo o grupo casi siempre
es hidrógeno que puede ser sacado
por la base, dejando detrás su par de electrones.
(c) La reacción es provocada por la acción de una base.
La base es, típicamente, un anión fuertemente
básico, como el hidróxido, o un
alcóxido derivado de un alcohol
(Sec. 5.5): etóxido, C2H5O-; t-butóxido, (CH3)3CO-;
etc. Pero, a veces, el propio disolvente, una sustancia neutra como un alcohol
o agua, sirve de base, aunque considerablemente más débil.
Por conveniencia, particularmente para
designar disolventes o reactivos, a menudo se abrevian los nombres de los
grupos alquilo más sencillos: metilo, Me; etilo, Et; n-propilo, n-Pr;
isopropilo, i-Pr; t-butilo, t-Bu. El metanol se convierte así en MeOH; el
metóxido de sodio, en NaOMe; el ion metóxido, en MeO-.
En la eliminación, un grupo saliente bueno es
un anión o una molécula débilmente básicos, igual que en la sustitución
nucleofílica y por las mismas razones-. Como base débil, libera un protón con
facilidad; como buen grupo saliente, libera carbono fácilmente (Sec. 5.9). En
la deshidrohalogenación, el grupo saliente es un ion halogenuro muy poco
básico; no es accidental que los halogenuros de alquilo sean sustratos
importantes, tanto en la sustitución nucleofílica como en la eliminación.
Tampoco es mera coincidencia que pueden
utilizarse las mismas alternativas para halogenuros de alquilo en ambas clases
de reacción otros sustratos capaces de liberar aniones débilmente básicos-.
Entre estos últimos, los más importantes son los tosilatos, que vimos en la sección 5.9, y sus allegados, otros
sulfonatos.
La similitud de lso sustratos en la
sustitución nucleofílica y en la eliminación, combinada con el hecho de que
tanto los nucleófililos, como las bases son reactivos ricos en electrones de
hecho, a menudo son el mismo reactivo-, puede conducir a problemas: siempre hay
la posibilidad de competencia entre
ambas reacciones (Sec. 7.24).
Pues bien, ¿qué mecanismo, o mecanismos,
sigue la deshidrogenación? Por el solo examen de las estructuras de los
reactivos y productos hemos llegado a ciertas conclusiones sobre lo que sucede
durante la reacción: se rompen los enlaces carbono-halógenoi y
carbono-hidrógeno; se forman los enlaces n y base-hidrógeno. No obstante, esta
descripción aún dista mucho de ser un mecanismo. Se rompen y forman enlaces, y
debemos plantear la pregunta: ¿Cómo depende del tiempo todas estas rupturas y formaciones de enlaces? ¿Suceden
todas al mismo tiempo o una tras otra? Y, es una después de otra, ¿Cuál ocurre primero?
En lo que concierne a la dependencia del
tiempo, hay varias posibilidades, cada una de ellas correspondiente a un
mecanismo diferente. En las secciones siguientes usaremos la prueba experimental la colección de los hechos para establecer cuál de estos
mecanismos en un determinado conjunto de condiciones sigue realmente la
eliminación. Sin embargo, veamos primero cuáles son estos posibles mecanismos,
para comprender mejor el significado de cada uno de los hechos que se presentan.
Debemos tener en cuenta que más importante que lo estudiado acerca de la
eliminación es conocer en profundidad algo de los métodos utilizados para
descubrir lo que sucede en una reacción química.
7.13 Cinética de la deshidrohalogenación.
Dualidad del mecanismo
La teoría de las reacciones de eliminación se
desarrolló de una forma notablemente similar a la de la sustitución
nucleofílica (Sec. 5.12). Otra vez en la década de 1930, se planteó una teoría
amplia de la reacción, y nuevamente se debió a Hughes e Ingold. También en este
caso propusieron dos mecanismos que difieren en su molecularidad. Mucho de lo
que trataremos más adelante se basa en el trabajo realizado desde sus
proposiciones iniciales y en la labor de otros investigadores. Este trabajo
posterior ha conducido a refinamientos de la teoría, y nos ha proporcionado una
visión más detallada de lo que sucede; sin embargo, en lo esencial, se ha
ajustado notablemente bien al esquema que desarrollan Hughes e Ingold.
Comencemos nuestro estudio donde lo hicieron
Hughes e Ingold: con la cinética de
la fuerte, la deshidrohalogenación sigue
una cinética de segundo orden; esto es, la velocidad de formación del
alqueno depende de la concentración de dos sustancias: el halogenuro de alquilo
y la base. Esta reacción de segundo orden se observa para todas las clases de
halogenuros de alquilo.
Velocidad
= k [ RX ] [:B]
Pues
bien, si se procede con una serie de sustratos, de 1º a 2º a 3º, y si se reduce la fuerza o la
concentración de la base, aparece un segundo tipo de comportamiento: cinética de primer orden. La velocidad
de la eliminación depende sólo de la concentración del halogenuro de alquilo y
es independiente de la concentración de la base.
velocidad
= K [RX]
En general, esta reacción de primer orden
solamente se encuentra en sustratos secundarios y terciarios, y en soluciones
donde la base es débil o se halla en concentración baja.
Para explicar estos dos tipos de
comportamiento cinético, Ingold y Hughes propusieron que la eliminación, al igual que la sustitución nucleofílica,
puede proceder por dos mecanismos
distintos. Por razones que mencionaremos luego, estos mecanismos se
denominan E2 y E1.
7.14 Mecanismo E2
Para la reacción que procede mediante una
cinética de segundo orden, Ingold y Hughes propusieron el mecanismo E2. La reacción implica un solo paso: la base arranca un protón del carbono; de modo
simultáneo se aleja un ion halogenuro y se forma el enlace doble. El halógeno
se lleva consigo su par de electrones, y el hidrógeno deja el suyo atrás, para que se forme el doble
enlace. Como ya dijimos, éstos son los cambios electrónicos que deben ocurrir
en la deshidrohalogenación; lo que caracteriza a este mecanismo específico, es
que todo sucede simultáneamente, en una sola etapa, mediante un solo estado de
transición.
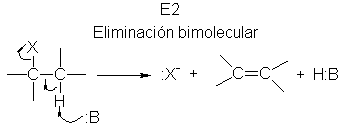
En este estado de transición se están
rompiendo dos enlaces: C-H y C-X. ¿De dónde procede la energía para estas
rupturas? Como es usual, procede de la formación
de enlaces: formación del enlace entre el protón y la base, y formación del
enalce n. (Aun cuando es más débil que el enalce o, el n proporciona unas 70
kcal/mol.)
Consideremos lo que sucede cuando la base
comienza a alejar el protón de la molécula.
El carbono ß, armado con el par de electrones
abandonado por el protón que se aleja, comienza a formar un enlace con
elcarbono a: un segundo enlace, el n. A medida
que comienza a formarse este último, se inicia la ruptura
carbono-halógeno. El halógeno está siendo empujando
hacia afuera en lo que, desde el punto de vista del carbono a, es una
especie de ataque nucleofílico, no muy distinto de una reacción SN2.
(Volveremos sobre este punto en la Sec. 9.7.)
El mecanismo descrito fue propuesto para la
eliminación de segundo orden. La cinética de segundo orden es exactamente, por
supuesto, lo que debe esperarse para una reacción que procede por el mecanismo
E2. El paso determinante de la velocidad la única
etapa
Velocidad = K [RX ] [:B]
Reacción E2
Cinética de segundo orden
Implica una reacción entre una molécula de
halogenuro de a alquilo y otra de una base, y su velocidad es proporcional a la
concentración de los reactivos. Este mecanismo se denominó E2, esto es, eliminación bimolecular, porque en el
paso que determina la velocidad, dos moléculas sufren cambios en su covalencia.
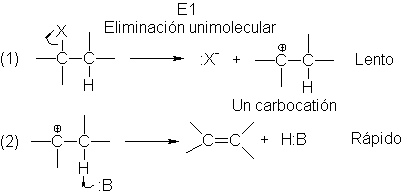
7.15 Mecanismo E1
Para la reacción que procede con cinética de
primer orden, Hughes e Ingold propusieron el mecanismo E1. En éste, los cambios electrónicos la ruptura y
formación de enlaces son los mismos que en E2, pero en este caso no suceden
simultánea, sino sucesivamente. Mientras que E2 implica un solo paso. El involucra dos. En el paso
(1) el sustrato sufre una heterólisis lenta para formar un ion halogenuro y un
carbocation. En el paso (2) el carbocatión pierde rápidamente un protón, que
pasa a la base, y genera el alqueno.
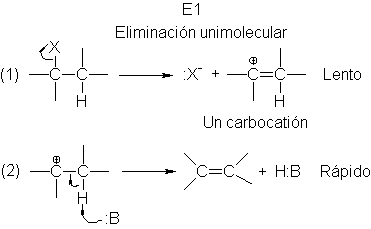
Reconocemos que el paso (1) es idéntico al
primer paso en SN1. En la segunda etapa de este último se combina el
carbocation con un nucleófilo para producir un compuesto de sustitución; en el
paso (2) de E1, el carbocatión reacciona con la base para dar el producto de la
eliminación.
Aquí, como siempre, las reacciones de los
carbocationes tienen un final común: proporcionan
un par de electrones para completar el octeto del carbono deficiente en
electrones. En la SN1, estos electrones son un par no compartido
del nucleófilo; en E1, se trata de un par compartido originalmente con el
proton que queda dsiponible mediante la formación de un enlace n- por la salida
del protón.
Basandonos en el paso (2, podemos agregar
otra reacción a nuestra lista de la sección 5.23.
Un
carbocatión puede:
(a) combinarse con un nucleófilo;
(b) transponerse a un carbocatión
más estable;
(c) eliminar un protón para generar al alqueno.
Esta lista seguirá creciendo (Sec. 8.16).
La reacción E1 sigue una cinética de primer
orden, igual que la SN1, y por exactamente la misma razón. La
velocidad global de la reacción queda determinada solamente por la primera
etapa que es lenta. Salvo por las numerosas moléculas de disolvente necesarias,
este paso determinante de la velocidad
sólo implica al sustrato, y su velocidad sólo depende de la concentración de
aquél. La velocidad de una reacción E1 es independiente de la concentración de
la base, porque la reacción cuya
velocidad estamos midiendo no implica base.
Nuevamente, la velocidad de la información de
los carbocationes es la que determina la rapidez de una reacción. Una vez
formados, los carbocationes reaccionan rápidamente para generar prodeucto; en
este caso, el alqueno.
Este mecanismo se denominó E1, esto es, eliminación unimolecular, porque en el
paso determinante de la velocidad, sólo una molécula, el sustrato, sufre un
cambio de covalencia.
7.16 Evidencia para el mecanismo E2. Ausencia de transposición
Veamos ahora las pruebas que indican cuál de
estos mecanismos se sigue realmente en un conjunto particular de condiciones.
Comencemos con el tipo de sistema ya descrito: la deshidrohalogenación con una
solución alcohólica concentrada de una base fuerte. En tales condiciones, la
reacción sigue una cinética de segundo orden.
Las reacciones de eliminación que
(a) siguen una cinética de
segundo orden,
también:
(b) no van acompañadas de
transposiciones;
(c) presentan un efecto
isotópico de hidrógeno importante;
(d) no van acompañadas de
intercambio de hidrógeno, y
(e) presentan un efecto
elemento grande.
El mecanismo consistente con estos hechos es
el E2, por lo que es generalmente aceptado para la reacción de segundo orden.
Es el más importante de los mecanimos descritos, puesto que es la ruta
principal seguida, no sólo por la deshidrohalogenación, sino por las
eliminaciones 1,2 en general. Veamos cada una de las evidencias: cómo sostienen
el mecanismo E2 y cómo ayudan a descartar las alternativas.
En primer lugar, estas reacciones (a) siguen una cinética de segundo orden. Ya
vimos (Sec. 7.14) que esto es consecuente con el mecanismo E2. No lo es con el
E1, que debe llevar a una cinética de primer orden.
A continuación, estas eliminaciones de
segundo orden (b) no van acompañadas de
transposiciones. También este hecho concuerda con el mecanismo E2, cuyo
paso único sencillamente no ofrece oportunidades para una transposición. A
igual que la cinética, este hecho es inconsistente con el E1, puesto que los
carbocationes, como ya sabemos, son muy propensos a la transposición (Sec.
5.23).
7.17 Evidencia para el mecanismo E2. Efectos isotópicos
Llegamos ahora a la tercera evidencia para el
mecanismo E2. Estas eliminaciones de segundo orden (c) presentan un efecto isotópico de hidrógeno importante. Para
comprender lo que esto significa, debemos aprender primero lo que es un efecto
isotópico y lo que significa en general.
Por definición, los diferentes isótopos de un
mismo elemento tienen la misma configuración electrónica y, por consiguiente,
propiedades químicas similares. Esta similitud es el fundamento de la técnica
de los trazadores isotópicos (Sec. 3.29): un isótopo hace lo mismo
prácticamente que otro, pero puede ser detectado en una secuencia química por
su radiactividad o por su masa inusual.
Puesto que los isótopos tienen, también por
definición, masas diferentes, sus propiedades químicas no son idénticas: las mismas reacciones se producen a una velocidad
algo diferente (o, para reacciones reversibles, con posiciones de equilibrio
distintas). Una diferencia en velocidad
(o en posición de equilibrio) debida a la presencia de un isótopo distinto en
el sistema de la reacción se llama efecto
isotópico.
Consideraciones teóricas que no podemos
tratar, apoyadas por muchas observaciones experimentales, conducen a la
conclusión siguiente: si un átomo
determinado se halla menos firmemente unido en el estado de transición de una
reacción que en el reactivo, la reacción que implica al isótopo más pesado de
dicho átomo procederá más lentamente. Los
isótopos del hidrógeno son los que tienen proporcionalmente la mayor
diferencia másica: el deuterio (D) es dos veces más pesado que el protio (H), y
el tritio (T), tres veces; lo que se traduce en que lso efectos isotópicos del
hidrógeno son más intensos, más fáciles de medir, y debido a la importancia
especial del hidrógeno en la química orgánica más estudiados. (Si el lector
duda de la importancia del hidrógeno, le bastará con observar la estructura de
prácticamente todos los compuestos orgánicos de este libro.)
Un tipo de reacción en la que el átomo está
menos firmemente unido en el estado de transición que en el reactivo, es un
proceso donde se está rompiendo un enlace con ese átomo. Los efefctos
isotópicos debido a la ruptura de un enalce con el átomo isotópico se conocen
como efectos isotópicos primarios,
que, en general, son los mayores que pueden observarse para un conjunto
determinado de isótopos.
En este libro nos ocuparemos de efectos isotopicos primarios de hidrógeno,
que equivalen a lo siguiente: Un enlace
de protio (H) se rompe más rápidamente que uno de deuterio (D).
Para muchas reacciones de este tipo.
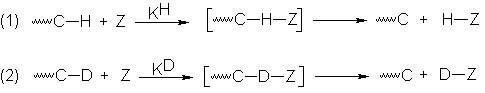
En las que se separa hidrógeno como átomo,
ion positivo o negativo, se han observado efectos isotópicos de deuterio (KH/KD)
hasta en el intervalo de 5 a 8 (a temperatura ambiente); esto es, la reacción
es de 5 a 8 veces más rápida para el hidrógeno ordinario que para el deuterio.
(Los efectos isotópicos del tritio, KH/K1, son casi dos veces mayores que los del
deuterio.)
Estas diferencias de velocidad se pueden
medir de diversas maneras. En algunos casos se puede medir directamente la
velocidad de cada reacción individual
(1) y (2) y comparar las constantes de velocidad KH y KD. Sin embargo, a menudo es más
factible el empleo de nuestro conocido método de competencia (Sec. 3.22) en
cualquiera de sus dos formas.
En la competencia intermolecular compite una mezcla de reactivos marcados y no
marcados por una cantidad limitada de un reactivo, de modo que las reacciones
(1) y (2 suceden en una misma mezcla; luego, medimos las cantidades relativas
de H-Z y D-Z que se producen. (A veces se emplean cantidades mayores del
reactivo Z y se determinan las cantidades relativas no consumidas de los dos reactivos, el ordinario y el marcado;
predominará el menos reactivo, por haber sido consumido más lentamente. La
velocidad relativa de cada reacción puede calcularse sin mayor dificultad.)
En la competencia intramolecular se emplea un solo reactivo que contiene varias
posiciones equivalentes, algunas marcadas y otras no:
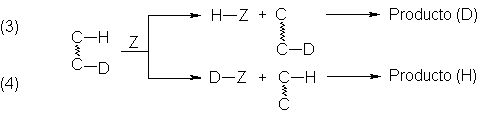
Luego es posible determinar bien las
cantidades relativas de H-Z y D-Z, o bien las cantidades relativas del producto
que contiene D, formado por la reacción (3), y del que contiene H, obtenido de
la reacción (4).
La presencia o ausencia de un efecto isotópico para una reacción en particular
puede tener una importancia enorme para el químico orgánico. Como primer
ejemplo de la utilización de este concepto, volvamos a nuestro tema original,
la evidencia que apoya el mecanismo E2.
Estudiemos el halogenuro de alquilo
sustituido, bromuro de 2-feniletilo, C6H5CH2CH2Br.
El grupo fenilo, C6H5,
se deriva del compuesto aromático benceno, C6H6. (A
menudo se representa el grupo fenilo por Ph.) Cuando se encuentra unido a un
halogenuro de alquilo, este grupo ejerce ciertos efectos sobre las reacciones
del halogenuro, incluyendo la eliminación: destacaremos estos efectos a medida
que nos encontremos con ellos, y los trataremos más adelante (Cap. 15). Pero
por el momento sólo es necesario saber
que el grupo C6H5 mismo es inerte frente a los reactivos que
ocasionan la eliminación, de modo que puede considerársele sólo como un
sustituyente más.
Se preparó bromuro de 2-feniletilo mercado, C6H5CD2CH2Br.
Este compuesto contiene deuterio en ambas posiciones ß, las posiciones en las
cuales debe perderse hidrógeno en una eliminación. Se determinó la constante de
velocidad (KD) para su deshidrobromación mediante etóxido de sodio,
y se la comparó con la constante (KH) para la reacción del bromuro
de 2-feniletilo ordinario (sin marcar) en iguales condiciones. Se halló que KH/KD=7,
esdecir, el compuesto que contiene protio reacciona siete veces más rápido que el que contiene deuterio. Vimos que un
efecto isotópico de esta magnitud es lo que se espera para la ruptura de un
enlace carbono-hidrógeno.
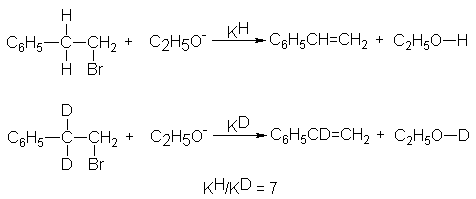
Ahora bien, ¿qué tiene de significativa aquí
la existencia de un efecto isotópico? No es que esté indicando la ruptura de un
enlace carbono-hidrógeno ß, puesto que esto ya lo sabemos, por los productos de
la reacción. Lo significativo es que indica la ruptura de un enlace
carbono-hidrógeno ß en un paso
determinante de la velocidad.
Este hecho es, por supuesto, consistente con
el mecanismo E2: se rompe un enlace carbono-hidrógeno ß en el único paso (y por
consiguiente determinante de la velocidad).
Para poder apreciar la importancia de esto,
consideremos lo que sería de esperar de una reacción que procediera para el mecanismo E1. También en este caso
se deshace un enlace carbono-hidrógeno ß, y se perdería más rápidamente protio
que deuterio pero del carbocatión, en el segundo paso, el rápido, cuya velocidad no tiene ningún efefcto sobre la
reacción total; la velocidad queda
determinada por la primera etapa, la formación del carbocatión, que no
involucra la ruptura de un enlace carbono-hidrógeno. (Habría un efecto
isotópico sobre este primer paso, pero sería secundario, mucho menor que
el primario observado para la reacción
E2.) La velocidad global de la reacción E1, medida por la desaparición del
sustrato, no indicaría ningún efecto isotópico primario.
7.18 Evidencia para el mecanismo E2.
Ausencia de intercambio de hidrógeno
Los hechos (a), (b) y (c) son, exactamente,
lo que esperaríamos para el mecanismo E2. El paso determinante de la velocidad
(el único paso), en el cual se rompe
un enlace carbono-hidrógeno y no existe la oportunidad para que ocurran
transposiciones, implica la reacción entre una molécula de halogenuro de
alquilo y una de base. Estos tres hechos, en particular, eliminan un mecanismo
de carbocationes (E1) para la eliminación de segundo orden.
Hasta este momento solamente hemos
considerado dos mecanismos para la deshidrohalogenación, uno, (E2), en el que
halógeno e hidrógeno abandonan el sustrato al mismo tiempo, y otro, (E1), en el
que sale primero el halógeno. Existe una tercera posibilidad razonable, un
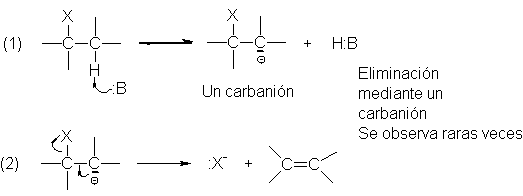
mecanismo en el que se va primero el hidrógeno: el mecanismo del carbanión.
Tal mecanismo tiene dos pasos, en el (1), el
sustrato pierde un protón por acción de la base para generar una partícula con
carga negativa: un carbanión. En el
paso (2), este carbanión pierde un ion halogenuro para dar el alqueno.
El paso (1) es una reacción ácido-base en el
sentido de Lowry-Bronsted, en el que el sustrato actúa como un ácido. Puesto que el protón ácido está unido a un
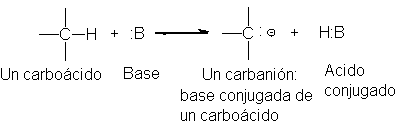
carbono, el sustrato se denomina carboácido,
el cual, como la mayoría de ellos, es extremadamente débil.
Los productos del paso (1) son el ácido
conjugado de la base por ejemplo, agua, de la base, ion hidróxido y la base
conjugada del carboácido, un carbanión. Un carbanión
es la base conjugada de un carboácido.
(La eliminación mediante carbaniones a menudo
se denomina E1Bc, eliminación
unimolecular de la base conjugada.)
Al igual que el mecanismo E2, el del
carbanión es congruente con los hechos (a), (b) y (c). Se han desarrollado
experimentos para distinguir entre estas dos posibilidades; en ellos se empleó
deuterio como marca, esta vez no para probar la acción de efectos isotópicos,
sino simplemente como trazador, para probar un eventual intercambio de hidrógeno. Veamos cómo opera esta idea.
Consideraremos la deshidrohalogenación del
bromuro de 2-feniletilo, C6H5CH2CH2Br.
(Se eligió este sustrato porque, como veremos (Sec. 15.17), el grupo fenilo
debería favorecer
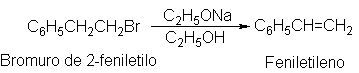
Notablemente la formación de carbaniones.) La
deshidrogenación se llevó a cabo con etóxido de sodio, C2H5Ona,
en solución de etanol. La formación de carbaniones implicaría la conversión de
la base, el ion etóxido, en su ácido conjugado, el etanol, que es el
disolvente.
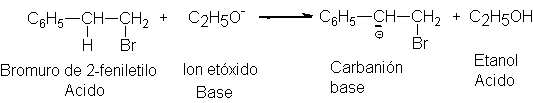
Pues bien, en el experimento real, el
sustrato fue bromuro de 2-feniletilo ordinario ( sin marca), y el disolvente
fue etanol marcado, C2H5OD.
Consideremos lo que sucedería, si se formaran carbaniones y se formaran
reversiblemente. Para regenerar material de partida, la mayoría de ellos
recuperaría hidrógeno muchas veces, antes de perder un ion halogenuro para dar
el alqueno. Y la recuperación de este hidrógeno vendría del disolvente, el ácido conjugado de la
base y, de hecho, el único ácido que puede encontrarse de acidez apreciable.
Pero casi todas las moléculas de disolvente son C2H5OD, y
no C2H5OH; de este modo, el carbanión casi con certeza
ganaría en esta reversión un deuterón,
en vez de un protón.
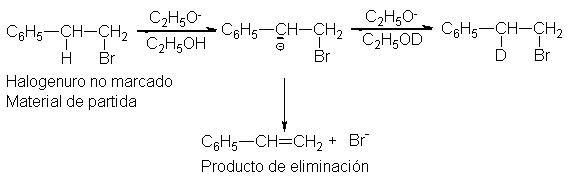
Se
permitió que la reacción procediera hasta la conversión de aproximadamente
la mitad del sustrato en alqueno. Luego, se interrumpió y se recuperó el
bromuro de 2-feniletilo no consumido. El análisis espectrométrico de masas
demostró ausencia de deuterio.
Experimentos similares con otros sistemas han
dado resultados semejantes. Las reacciones de eliminación de segundo orden
típicas (d) no van acompañadas de
intercambio de hidrógeno.
De esta manera, el hecho (d) descarta el
mecanismo con formación reversible de carbaniones. Concuerda, así pues, son el
mecanismo E2, que no da oportunidad para intercambio de hidrógeno.
7.19 Evidencia para el mecanismo E2. El efecto elemento
Estas eliminaciones de segundo orden (e)
presentan un efecto elemento grande.
Observemos una vez más los dos pasos del
mecanismo carbaniónico. La ausencia del intercambio de hidrógeno tratado antes
no descarta del todo tal mecanismo: simplemente indica que, si se forman carbaniones, lo hacen irresistiblemente; o sea, pierden iones
halogenuro mucho más rápido de lo que recobran protones. Esto es, K2
tendría que ser mucho mayor que K-1.
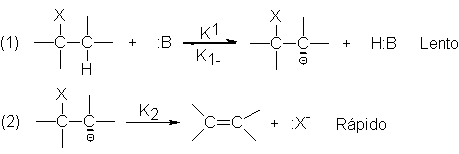
De ser así, el paso (1) sería
determinantemente de la velocidad y la velocidad del paso (2) no tendría ningún
efecto sobre la velocidad total de la reacción como en una reacción SN1
o E1-. Según sean las condiciones, el paso (2) podría se más rápido o más
lento, pero realmente esto no importa: el paso (1) formaría un cuello de
botella, y su velocidad determinaría lo rápida que es la eliminación.
En esta altura de nuestro análisis nos hemos
quedado con dos mecanismos posibles para la eliminación de segundo orden: el E2
y el carbaniónico, cuya etapa determinante de la velocidad es la formación del
carbanión. Para elegir entre ambos, debemos reconocer esta diferencia crucial:
en la etapa que determina la velocidad de E2, se está rompiendo el enlace
carbono-halógeno, que no es el caso en el mecanismo carbaniónico. La facilidad
de la ruptura del enlace carbono-halógeno debería, por tanto, afectar a la
velocidad de la eliminación de E2, pero no a la del mecanismo carbaniónico
(irreversible).
Ahora, para establecer si un enlace determinado se está rompiendo o no
en la etapa determinante de la velocidad, podríamos considerar la búsqueda de
un efecto isotópico, como se hizo en el estudio de la ruptura del enlace
carbono-hidrógeno (sec. 7.17), tarea más difícil ahora. No estamos tratando con
la pérdida de hidrógeno, cuyos isótopos difieren dos y tres veces en masa, sino
con la pérdida de elemento más pesados, como el cloro, cuyos isótopos sólo
difieren en un porcentaje pequeño, con las diferencias correspondientemente
pequeñas en la facilidad de ruptura de los enlaces.
Joseph Bunnett (Universal de California,
Santa Cruz) ha señalado, al referirse a este punto, que existe evidencia de lo
que él denominó el efecto elemento
desde hace muchos años.
Las energías de disociación heterolítica de
enlaces (Tabla 1.3, Sec. 1.14) indican que la fuerza de los enlaces
carbono-halógeno sigue la secuencia
![]() Energía de disociación
heterolítica de enlace
Energía de disociación
heterolítica de enlace
En las reacciones SN2 y SN1
el enlace carbono-halógeno se rompe en el paso determinante de la velocidad.
Por otra parte, la reactividad en la sustitución nucleofílica sigue la
secuencia,
![]()
Reactividad
en SN2 o SN1
En que la velocidad de la reacción refleja la
facilidad de ruptura del enlace carbono-halógeno.
Aquí, las diferencias en velocidad son
importantes; por ejemplo, lso bromuros de alquilo reaccionan de 25 a 50 veces
más velozmente que los cloruros correspondientes. De hecho, los efectos
elemento son mucho mayores que los
isotópicos observados para la ruptura de uniones con protio y deuterio;
como debe ser, por cierto, en
consideración a las diferencias mucho más grandes en energías de enlace.
Ahora bien, en estas reacciones de
eliminación las reactividades de los halogenuros de alquilo siguen la misma
secuencia que para la sustitución.
![]()
Reactividad en E2
Y con efectos elemento de la misma magnitud
aproximada: los bromuros de alquilo reaccionan entre 40 y 60 veces más
velozmente que los cloruros y para comparar el intervalo completo de la reactividad
los yoduros lo hacen por encima de 25 000 veces más rápido que los fluoruros.
Por esta razón, la velocidad de ruptura
del enalce carbono-halógeno afecta a
la velocidad global de la eliminación.
De hecho, se han medido efectos isotópicos de
grupos salientes para eliminaciones que resultaron ser de magnitud consecuente
con una ruptura de enlace considerable en el estado de transición.
Por consiguiente, sólo el mecanismo E2 se
ajusta a todos los hechos, por lo que es generalmente aceptado como la vía
principal de la mayoría de las eliminaciones 1,2.
7.20 Reacción E2: orientación y reactividad
Hasta ahora hemos tratado la evidencia de que
la deshidrohalogenación procede mediante el mecanismo E2. Veamos algunas otras
características.
Dijimos (Sec. 7.12) que la
deshidrohalogenación da a menudo una mezcla de alquenos isómeros. ¿Qué isómeros
predominará en tal caso? El análisis de muchas reacciones ha indicado que por
lo general predomina uno, y que es posible predecir cuál será esto es, predecir
la orientación de la eliminación
basándonos en la estructura molecular.
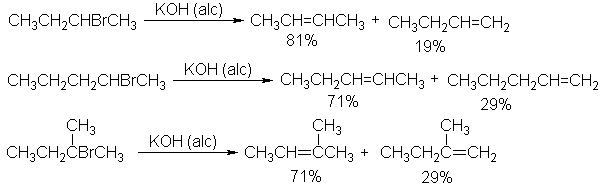
Por ejemplo, tomemos, el bromuro de sec-butilo. El ataque por medio de una
base a cualquiera de los hidrógenos ß (los del C-1) puede llevar a la formación
de !-buteno; el ataque a cualquiera de los hidrógenos ß (del C-3) puede dar
origen al 2-buteno. Apreciamos que el producto preferido es el 2-buteno, a pesar del factor de probabilidad 3:2 que
opera en su contra.
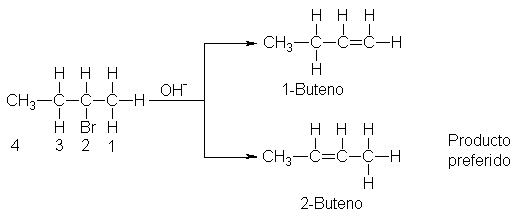
Si concentramos nuestra atención en el
alqueno que se está formando en lugar de hacerlo en el hidrógeno que se pierde,
observaremos lo siguiente: el producto preferido, 2-buteno, es un alqueno disustituido, mientras que 1-butano es monosustituido; es decir, en el 2-buteno
hay dos grupos alquilo (dos grupos CH3) ligados a los carbonos del
doble enlace, mientras que en el 1-buteno sólo hay uno (C2H5).

En otros ejemplos onservamos que se prefiere
un alqueno disustituido a uno mono-sustituido, y uno con tres sustituyentes, a
uno con dos.

Estos forman parte de una norma que observó
por primera vez el químico ruso Alexander Saytzeff (Universidad de Kazan),
quien formuló en 1875 una <<regla>> que puede resumirse así: el producto preferido en una
deshidrohalogenación es el alqueno que tiene el número mayor de grupos alquilo
ligados a lso carbonos con doble enalce.
Ahora bien, la deshidrohalogenación es una
reacción irreversible, de modo que, una vez más, la orientación queda
determinada por la velocidad relativa de reacciones que compiten.
Partiendo del bromuro de sec-butilo, se
obtiene más 2- que 1-buteno, porque el primero se forma más velosmente que el
segundo. El alqueno con el número más grande de grupos alquilo es el producto
de preferencia, porque se genera con mayor velocidad que los alquenos alternativos.
La regla de Saytzeff proporciona así una secuencia que indica la velocidad
relativa de formación de alquenos.
Facilidad de
formación de alquenos
![]()
En la sección 8.4 descubriremos pruebas de
que la estabilidad de los alquenos se ciñe exactamente a la misma secuencia.
Estabilidad de
alquenos
![]()
Partiendo de esto, podemos reformular la regla de Saytzeff para que exprese lo
siguiente: en la deshidrohalogenación,
cuanto más estable es un alqueno, más velozmente se generará. La formación
predominante del isómero más estable se llama orientación de Saytzeff.
De esta forma, la regla es de utilidad más
general, puesto que se aplica a casos
en que la estabilidad de los alquenos se determina por características
estructurales distintas de los sustituyentes alquílicos (Secs. 10.12 y 15.19).
Además, esta formulación conduce directamente al factor que de hecho opera.
Consideraremos el estado de transición para
la reacción E2. Los enlaces del hidrógeno y del grupo saliente están
parcialmente rotos, mientras que el doble enlace está parcialmente formado. Por
tanto, el estado de transición adquiere un carácter
de alqueno considerable.
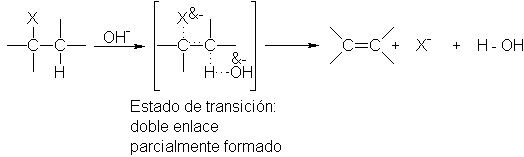
Los factores que estabilzian el alqueno en
este caso, grupos alquilo también estabilizan el alqueno incipiente en el
estado de transición. Disminuye la Eact, y el alqueno se genera más
velozmente. Una vez más, como en la formación de radicales libres y de
carbocationes, el carácter de producto del estado de transición es un factor
importante en la determinación de su estabilidad, y por consiguiente, de su
velocidad de reacción.
Sin embargo, el carácter de alqueno del
estado de transición no es el único factor que opera en la eliminación; de
donde resulta que la orientación no es siempre Saytzeff. Esto se cumple
especialmente cuando están involucrados sustratos distintos de halogenuros o
sulfonatos de alquilo. En la sección 27.6 estudiaremos otro tipo de
orientación, la de Hofmann, además de
lso factores que la provocan. Reconoceremos que la orientación en la
eliminación es el resultado neto de la operación de varios factores que a
menudo se oponen y que la orientación de Saytzeff, que por lo general se
observa para las eliminaciones en ahlogenuros y sulfonatos de alquilo, refleja
un estado de transición en el cual
domina un factor: la estabilidad del alqueno.
La estabilidad del alqueno no sólo fija la orientación de la deshidrohalogenación,
sino que también es un factor importante en la determinación de la reactividad de un halogenuro de alquilo
en la eliminación, como se muestra más adelante, por ejemplo, para la reacicón
con etóxido de sodio en etanol a 55ºC. Vemos que, incluso después de considerar
el número de hidrógenos ß, la velocidad relativa por hidrógeno aumenta con la mayor sustitución del alqueno.

Por definición, a medida que uno procede en
la serie de halogenuros de alquilo de 1º a 2º a 3º, la estructura se vuelve más
ramificada en el carbono portador del halógeno. Esta ramificación creciente
arroja dos resultados: proporciona un número mayor de hidrógenos B para el
ataque mediante una base y, en
consecuencia, un factor de probabilidad más favorable para la eliminación, y
además conduce a un alqueno más ramificado, más estable y, por consiguiente, a
un estado de transición más estable y a una Eact más baja. Como resultado de
esta combinación de factores, en una
deshidrogenación E2 el orden de reactividad de los halogenuros de alquilo es
![]()
Reactividad de RX en E2
No obstante, podemos profundizar aún más en
el análisis de la estructura de los sustratos. Un sustrato puede ser de la
misma clase que otro, y sin embargo, dar un alqueno más ramificado, y, en
general, esperaremos que sea más reactivo. Usualmente, esto es cierto, aunque
el número de hidrógenos ß sea menor; donde ambos factores se oponen, la estabilidad
del alqueno tiende a pesar más que el factor de probabilidad.
En el capítulo o examinaremos un aspecto más
de la reacción E2: su estereoquímica
(Sec. 9.7).
7.21 Evidencia para el mecanismo E1
Hasta este momento se ha puesto énfasis en
E2, el mecanismo que Hughes e Ingold propusieron para la deshidrohalogenación
con una cinética de segundo orden. Sin embargo, la deshidrohalogenación también
puede proceder con cinética de primer orden. Para esta reacción, Ingold y
Hughes propusieron otro mecanismo, el E1.
Vimos que este mecanismo implica dos pasos
(Sec. 7.15): una heterólsisi lenta del halogenuro de alquilo (paso 1) para
generar el arbocatión, que pierde rápidamente un protón que va a la base (paso
2) para dar el alqueno.
La reacción E1 sigue una cinética de primer
orden, porque el apso (1) es determinante de la velocidad, y la velcoidad de
este paso depende sólo de la concentración sustrato. La velocidad total es independiente de la
[base], puesto que no entra en la reacción hasta el segundo paso, el rápido.
![]()
Pero consideraremos una situación en la que
es el propio disolvente el que lleva a
cabo la eliminación, en lugar de una base agregada: digamos, mediante etanol.
Según indica la cinética, la reacción podría
estar ocurriendo por el mecanismo E2, en el que el disolvente estaría
extrayendo un protón en el paso único, determinante de la velocidad. Sin
embargo, la
![]()
Cinética no revelaría esto: la concentración
del disolvente no varía durante la reacción, por lo que observaríamos una cinéticade
primer orden una cinética de seudoprimer orden, pero imposible de distinguir de la primer orden verdadero-. (La
constante de velocidad observada, Kobs, sería de hechouna constante
de velocidad verdadera, multiplicada
por la concentración del disolvente.) La situación es análoga a la de la
solvólisis (Sec. 6.9); de hecho, en condiciones solvóticas se sigue más
comúnmente el mecanismo E1. Por razones obvias, en esta situación se necesita
otro tipo de evidencia.
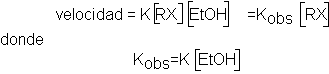
Pues bien, aparte de la cinética, ¿cuál es la
evidencia para el mecanismo E1? Las reacciones de eliminacion que
(a) siguen una cinética de
primer orden,
también
(b) no van acompañadas de un
efecto isotópico primario de hidrógeno;
(c) presentan el mismo efecto de la
estructura sobre la reactividad que las reacciones SN1, y
(d) van acompañadas de
transposiciones, donde la estructura lo
permite.
Examinemos cada uno de estos puntos.
Estas reacicones de primer orden (b) no van
acompañadas de un efecto isotópico
primario de hidrógeno. Vimos que tal efecto sólo es de esperar en una
eliminación, donde el enalce carbono hidrógeno-ß se rompe en el paso determinante de la velocidad (Sec. 7.17). Se
espera en E2, con su único paso. Se advierte un efecto isotópico primario
grande en las eliminaciones de segundo orden. No se espera observarlas en E1,
donde se pierde el protón en el segundo paso, rápido (véase Sec. 7.15). Esta es
la razón por la que no se contempla un efecto isotópico primario en las
eliminaciones de primer orden.
Luego, las eliminaciones de primer orden (c)
presentan el mismo efecto de la
estructura sobre la reactividad que las
reacciones SN1. Para comprender esta evidencia, debemos
recordar algo que ya hemos reconocido antes (Sec. 7.15): que E1 implica exactamente el mismo primer paso que SN1.
![]() Puesto que este primer paso es el que determina la velocidad, se
deduce que el orden de reactividad de
los halogenuros de alquilo en E1 debe
ser el mismo que en SN1. Experimentalmente, se ha demostrado que
esto es así.
Puesto que este primer paso es el que determina la velocidad, se
deduce que el orden de reactividad de
los halogenuros de alquilo en E1 debe
ser el mismo que en SN1. Experimentalmente, se ha demostrado que
esto es así.
Reactividad
en E1
La reactividad en E1, como en SN1,
se determina por la velocidad de la formación del carbocatión; y esto depende,
como ya vimos (Sec. 5.22), de la estabilidad del último.
Donde la estructura lo permite, estas
eliminaciones de primer orden van acompañadas de transposiciones.
Nuevamente volvemos al hecho de que el primer paso es el mismo que para SN1.
Por generarse carbocationes en esta primera etapa, se deduce que E1 debería ser
susceptible de transposiciones, y exactamente del mismo tipo de aquelals que
son características de SN1 (Sec. 5.23). Esto también se confirma
experimentalmente. El doble enlace aparece en lugares remotos con respecto al
carbono que estaba unido al grupo saliente:
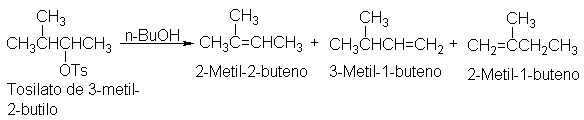
A veces se modifica el esqueleto carbonado:
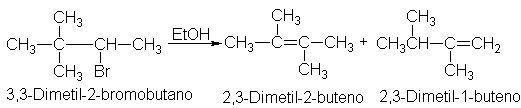
Los alquenos se obtienen incluso de sustratos
que no contienen hidrógeno ß:
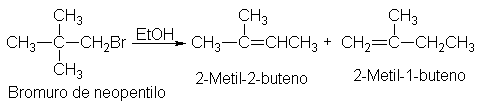
En cada caso, es evidente que si
efectivamente el alqueno se forma de un carbocatión, éste no es el mismo que se generó inicialmente del sustrato. Y, por
supuesto, no lo es.
En cada uno de estos ejemplos, el carbocatión
inicialmente formado se puede transponer mediante un desplazamiento 1,2 para
dar otro más estable. Como dijimos para las reacciones SN1 (Sec.
5.23), cuando esto puede suceder, sucede:
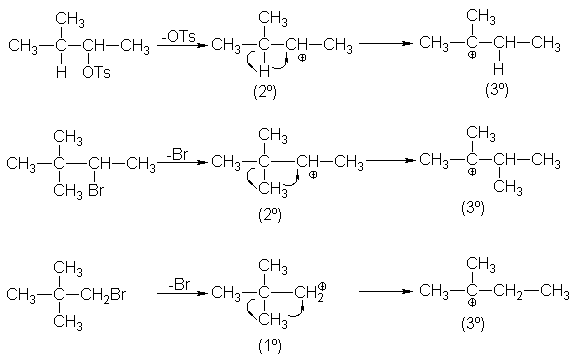
Este nuevo carbocatión es el que pierde el
protón de una manera directa desde una posición ß para dar origen a los
alquenos <<inesperados>>.
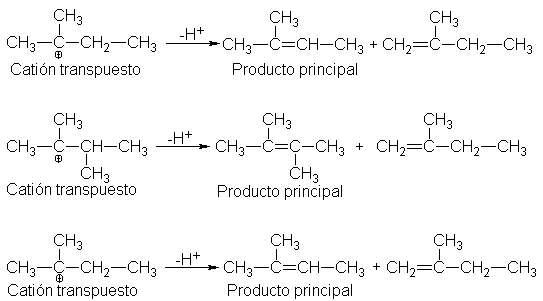
A partir de esto comenzamos a entender el
esuqema de transposiciones dado por reacciones de muchos tipos diferentes, el
patrón dado por primera vez por Meerwein en 1922
(Sec.
5.17), y que lo llevó a concebir el carbocatión como intermediario reactivo.
7.22 Reaccción E1: orientación
La eliminación por E1 muestra una fuerte
orientación de Saytzeff; esto es, cuando puede generarse más de un alqueno, el
producto preferido es el más ramificado el más
estable.
Así un alqueno disustituido es preferido a
uno monosustituido,
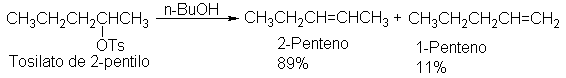
Y uno trisustituido es preferido a uno
disustituido.
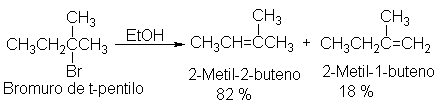
¿Cómo explicamos este tipo de orientación? En
otras reacciones estudiadas, la orientación y reactividad iban emparejadas.
Ambas están determinadas por velocidades de reacción relativas, y en el mismo paso: digamos, separación de
un hidrógeno por un átomo de cloro, o formación de un doble enlace una pérdida
concertada de un protón y del grupo saliente.
Sin embargo, en E1 encontramos una
diferencia. La orientación y la reactividad siguen estando determinadas por la
velocidad relativa de reacciones pero en
etapas diferentes.
La velocidad de reacción del sustrato está
determinada por la velocidad del paso (1). No obstante, el alqueno producido
está determinado por el protón ß que se pierde más rápidamente por el
carbocatión en el paso (2). El catión 2-pentilo, por ejemplo, puede perder un
protón del C-3 para generar 2-penteno, o del C-1 para dar 1-penteno. Hay
competencia, obteniéndose más 2-penteno, porque éste se forma más rápidamente.
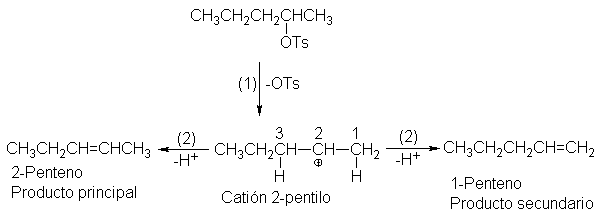
Examinemos el estado de transición para este
paso determinante del producto. El enlace carbono-hidrógeno está parcialmente
rotó, y el doble enlace está parcialmente formado.
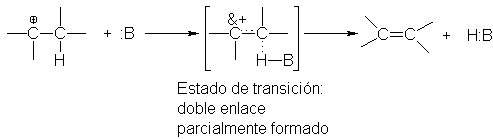
El estado de transición adquiere carácter alquénico. Como en E2, aquellos factores que estabilizan al
alqueno, también lo harán con el alqueno incipiente en el estado de transición.
Se rebaja Eact, y el alqueno se
forma con mayor velocidad.
Cuando ocurre una transposición en E1,
predecimos de cualquier manera una orientación de Saytzeff; sin embargo,
debemos considerar ahora la pérdida de protones ß, no sólo de los cationes
formados inicialmente, sino también de los transpuestos.
7.23 Eliminación: E2 contra E1
¿Cómo podemos anticipar que E2 o E1 es el
mecanismo que probablemente operará en un conjunto determinado de condiciones?
En primer luegar, observamos el efecto de la
naturaleza del grupo alquilo del sustrato. Al proceder a lo largo de la
secuencia 1º, 2º, 3º, auemnta la reactividad en ambos mecanismos, aunque por
razones diferentes, la reactividad por E2 aumenta principalmente porque se
forman los alquenos más ramificados, los más estables. La reactividad por E1
aumenta debido a la mayor estabilidad de losc arbocationes que se forman en el
paso determinante de la velocidad. En consecuencia, no podemos anticipar un
desplazamiento abrupto en el mecanismo por cambios simples en el grupo alquilo,
excepto para sustratos primarios, donde es muy difícil formar siguiera
carbocationes.
Pero si nos ocupamos de la función del otro
reactivo, la base, encontraremos una diferencia notable entre ambos mecanismos: en el E2, la base participa en
el paso determinante de la velocidad; en el E1, en cambio, no lo hace. [Ya hemos estudiado una competencia análoga
(Sec. 5.24) entre un mecanismo bimolecular (SN2) y uno monomolecular (SN1). Por
esto, lo que sigue no será una sorpresa.]
La velocidad de E2 depende de la concentración de la base, pero no la
de E1. La velocidad de E2 depende de la naturaleza de la base: una base más
fuerte extrae más rápidamente un protón del sustrato. La velocidad de E1es
independiente de la naturaleza de la base; más fuerte o más débil, la base
espera hasta que se haya formado el carbocatión.
Por tanto, para un sustrato determinado,
cuanto mayor sea la conentración o la fuerza de una base, más se favorece E2
sobre E1. En las condiciones que se utilizan típicamente para llevar a cabo una
deshidrogenación una solución concentrada de una base, la eliminación sigue el
mecanismo E2. En general, el mecanismo E1 se encuentra sólo con sustratos
secundarios o terciarios, y en soluciones donde la base tiene una concentración
baja o débil; por lo general, donde la base es el disolvente.
7. 24 Eliminación contra sustitución
Hemos dicho que los sustratos de uso más
corriente para una eliminación 1,2 promovida por bases, son halogenuros y
sulfonatos de alquilo. Estos compuestos son los mismos que actúan como
sustratos en las sustituciones nucleofílicas, y por una buena razón: ambas
reacicones precisan sustratos con buenos grupos salientes. Además, los
reactivos requeridos para ambos tipos de reacciones, bases y nucleofílos, son
similares; de hecho, a menudo son los mismos reactivos. Restos reactivos son
ricos en electrones; las bases son nucleofílicas, y los nucleófilos, básicos.
Resulta, por tanto, que, al menos en
principio, casi siempre habrá competencia
entre sustitución y eliminación.
Consideremos primero las reacciones
bimoleculares SN2 y E2. Dichas reacciones resultan del ataque al
sustrato por medio de un reactivo :Z. Actuando como nucleófilo, ataca un
carbono para llevar a cabo una sustitución; actuando como base, ataca un hidrógeno
para realizar una eliminación.
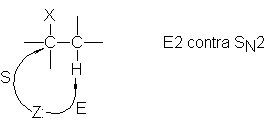
Entre sustratos, hemos visto que el orden de
reactividad es 3º > 2º > 1º
en E2. Para SN2, recordemos que el orden es el opuesto (Sec.
5.15). Al proceder a lo largo de la serie 1º, 2º, 3º, aumenta, por tanto, la
reactividad por E2, y disminuye por SN2.
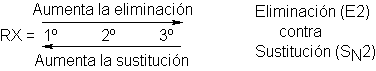
Los sustratos primarios son más lentos en la
eliminación y más rápidos en la sustitución; los terciarios son más rápidos en
la eliminación y más lentos en la sustitución. Cuando la sustitución y eliminación bimoleculares son reacciones que
compiten, crece la proporción de eliminación a medida que la estructura del
sustrato se modifica de primaria a secundaria, y de ésta, a terciaria.
Muchos sustratos terciarios producen en estas condiciones casi exclusivamente
alquenos.
La naturaleza del grupo alquilo es, quizás,
el factor principal que influye en la competencia entre SN2 y E2,
pero hay otros. Muchos nucleófilos, como el ion hidróxido, son bases fuertes,
de modo que la eliminación compite fuertemente con la sustitución. No onstante,
existen algunos reactivos que son buenos nucleófilos, pero bases relativamente
débiles, y con ellos tiende a favorecerse la sustitución.
Un disolvente menos polar tiende a favorecer
la eliminación, lo mismo que hace una temperatura más elevada. Por eso, el KOH
alcohólico caliente es el reactivo clásico para la deshidrohalogenación; una
temperatura más baja y la presencia de agua, más polar, en el disolvente
tienden a incrementar la proporción del producto de la sustitución: el alcohol.
Tratemos ahora la competencia entre las
reacciones unimoleculares, SN1 y E1. Como vimos, las dos tienen el
mismo paso, la heterólisis para la formación del carbocatión.
En la segunda etapa se bifurca la vía de la
reacción: una rama conduce a la sustitución,
y la otra, a la eliminación.
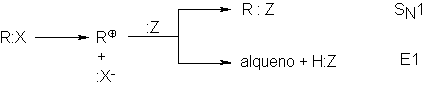
 En este segundo apso hay un ataque por el reactivo nucleofílico,
reactivo básico :Z, que típicamente es el disolvente. Esta vez, el ataque no va dirigido al propio sustrato,
sino al carbocatión. El ataque sobre carbono causa sustitución; el ataque sobre hidrógeno causa eliminación.
En este segundo apso hay un ataque por el reactivo nucleofílico,
reactivo básico :Z, que típicamente es el disolvente. Esta vez, el ataque no va dirigido al propio sustrato,
sino al carbocatión. El ataque sobre carbono causa sustitución; el ataque sobre hidrógeno causa eliminación.
Ahora bien, las proporciones de los productos
obtenidos cuánto producto de sustitución y cuánto alqueno están determinadas
por la velocidad relativa de estos segundos pasos alternativos. Como ya hemos
visto (Sec. 7.22), la velocidad con que el carbocatión pierde un protón depende
de la estabilidad del alqueno que se está formando para alquenos simpels, lo
ramificado que estén. Por tanto, la velocidad de la eliminaciónde un carbocatión
sigue la secuencia 3º > 2º > 1º. Es de esperar que la velocidad de la
sustitución la combinación con un nucleófilo siga la secuencia inversa, 1º >
2º > 3º, donde el menos estable es el de vida más breve. (De hecho, como
hemos visto en la Sección 6.9, incluso la heterólsiis de sustratos secundarios
generalmente implica la ayuda nucleofílica del disolvente. El catión formado
tiene en la parte de atrás una molécula de disolvente; hasta cierto punto, la
sustitución nucleofílica ya ha comenzado.)
Los hechos concuerdan con nuestro análisis:
en las reacciones unimoleculares, los sustratos terciarios dan la proporción
más elevada de eliminación. Por ejemplo, en etanol acuoso a 80ºC, el bromuro de
t-butilo da 19% del alqueno, mientras que el bromuro de isopropilo sólo da 5%.
Por tanto, nos enfrentamos a lo siguiente:
cuando queremos el producto de una reacción de sustitución, la eliminación es
un estorbo que debe ser envitado, pero no siempre puede lograrse. Con algunos
nucleófilos, resulta que sólo es posible lograr un rendimientoaceptable con
sustratos primarios, y posiblemente secundarios, y que los terciarios dan
virtualmente eliminación pura. Pero si queremos un alqueno, tratamos por todos los medios de lograr la
eliminación, para lo cual generalmente intentamos conducir la reacción hacia la
eliminación bimolecular: utilizamos un disolvente de polaridad baja y una
concentración elevada de una base fuerte.
7.25 Deshidratación de alcoholes
Hasta aquí hemos estudiado el tipo de
eliminación 1,2 promovido por una base. Analicemos ahora una eliminación
1,2 que es catalizada por un ácido: la deshidratación de alcoholes. A
pesar del cambio drástico en las condiciones de reacción, veremos que la
deshidratación no es muy distinta de la eliminación ya tratada.
Un alcohol se convierte en un alqueno por deshidratación: la
eliminación de una molécula de agua.
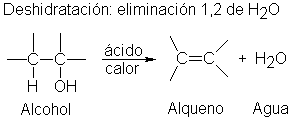
La deshidratación requiere la presencia de un
ácido y calor. En general, se puede proceder por dos métodos: (a) calentado el
alcohól con ácido sulfúrico o fosfórico, y (b) haciendo pasa el vapor del
alcohol sobre un catalizador, normalmente alúmina (Al2O3),
a temperaturas elevadas. (La alúmina funciona como un ácido, como un ácido
deLewis o, por medio de grupos OH en su superficie, como un ácido de Lowry-Bronsted.)
La facilidad de deshidratación de los
distintos tipos de alcoholes difiere mucho, siendo el orden de reactividades
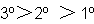 Facilidad de deshidratación de alcoholes
Facilidad de deshidratación de alcoholes
Los siguientes ejemplos ilustran cómo afectan
estas diferencias de reactividad a las condiciones experimentales de la
hidratación. (Ciertos alcoholes terciarios son tan propensos a la
deshidratación que sólo son destilables si se toman precauciones para proteger
el sistema de los vapores ácidos presentes en un laboratorio común.)
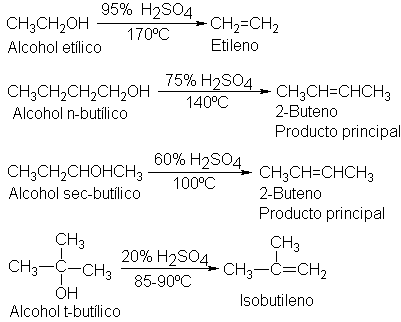
Para la deshidratación de alcoholes
secundarios y terciarios, generalmente se acepta el siguiente mecanismo.
El paso (1) es una reacción ácido-base
rápida entre el alcohol y el ácido catalizador que da el alcohol protonado y la
base conjugada del ácido. En el paso (2), el alcohol protonado sufre una
heterólisis para generar el carbocatión y agua. En el paso (3), el carbocatión
cede protón a la base para dar el alqueno.
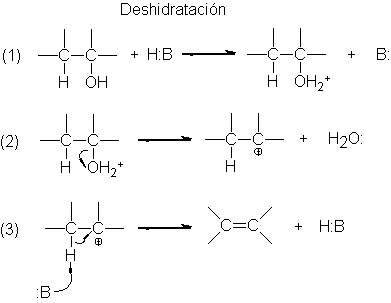
En los pasos (2) y (3) de este mecanismo
reconocemos una especie de leiminación E1 con el alcohol protonado como
sustrato. El paso (1), es sencillamente el preludio rápido y reversible que
produce el verdadero sustrato.
Veamos los hechos de la deshidratación e
intentemos explicar por medio de este mecanismo.
La
deshidratación es catalizada por ácidos. Se necesita un ácido para convertir el alcohol en la especie
protonada que puede sufrir la heterólisis para perder la molécula de agua
débilemnet básica. En consecuencia de ácido, la heterólisis requeriría la
pérdida del ion hidróxido fuertemente básico: un proceso que, como ya hemos
visto (Sec. 5.25), es tan difícil que raras veces, si alquna, sucede. El ácido
transforma el pésimo grupo saliente, OH, en uno excelente, -OH2+.
Dijimos que la deshidrohalogenación es promovida por bases: la reacción consume
base, por lo que debe estar presente en cantidades molares. Decimos que la
deshidratación es catalizada por
ácidos; no se consume ácido y, para los alcoholes más reactivos, sólo se
necesita la presencia de vestigios. Este hecho concuerda con el meanismo: el
ácido utilizado en el paso (1) es regenerado en el (3). Como ejemplo, tomemos
la deshidratación en ácido sulfúrico acuoso. El ion hidronio, H3O+,
es el ácido H:B; la base conjugada :B es agua. En el paso (1), el H3O+
pierde un protón para formar H2O; en el paso (3), H2O es
la base que recibe un protón del carbocatión, y al hacerlo se reconvierte en H3O+.
Observamos aquí una similitud fundamental
entre deshidratación y deshidrohalogenación. Una vez protonado el alcohol, lo
que requiere un medio ácido, una base desempeña su papel esencial acostumbrado
en la eliminación absorbiendo un protón.
La
deshidrtatción es reversible. Al contrario de
lo que ocurre con una eliminación 1,2 promovida por una base, ésta es reversible.
Como veremos más adelante, un ácido
cataliza la hidratación de alquenos para dar alcoholes. De acuerdo con esto, se
muestra cada paso del mecanismo como reversible. En condiciones de
deshidratación, el alqueno, que es bastante volátil, generalmente se desprende
de la mezcla de reacción, por lo que el equilibrio (3) es desplazado hacia la
derecha y, en consecuencia, toda la secuencia de reacciones es forzada hacia la
eliminación.
Ahora bien, de acuerdo con el principio de la reversibilidad microscópica,
una reacción y su inversa recorren
exactamente el mismo camino, pero en direcciones opuestas. (El paso más bajo en
una cordillera por un lado, también es el más bajo por el otro.) Partiendo de
esto, la deshidratación de alquenos. Por tanto, toda evidencia reunida acerca
del mecanismo de la hidratación y hay bastante (Secs. 8.9 a 8.12), se suma a
nuestra comprendión del mecanismo de deshidratación.
Hemos visto que el orden de reactividad de los alcoholes frente a la deshidratación es
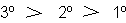 Facultad de deshidratación de alcoholes
Facultad de deshidratación de alcoholes
Hay evidencia (alguna procedente del estudio de la hidratación) de que la
velocidad de la deshidratación depende del paso (2), la formación del
carbocatión, y del (3), su pérdida de un protón. Los alcoholes terciarios se
deshidratan más rápido que los otros, porque forman los carbocationes más
estables, que una vez formados, estos
cationes dan los alquenos más estables.
En consecuencia, en un sentido estricto, la
deshidratación no es una reacción E1 del alcohol protonado.
En una eliminación E1 verdadera, la velocidad
de la reacción depende únicamente del paso de la heterólisis, ya que todo
carbocatión generado pasa de inmediato a producto; esto es, la pérdida de un
protón es mucho más veloz que la regeneración del sustrato. Este no es nuestro
caso: los carbocationes se forman reversiblemente a partir del alcohol
protonado y, de vez en cuando, uno de ellos pierde un protón para dar un
alqueno.
Donde lo permite la estructura del grupo
alquilo, hay transposición, que
sigue el esuqema observado para la deshidrohalogenación E1 (Sec. 7.21). Por
ejemplo:
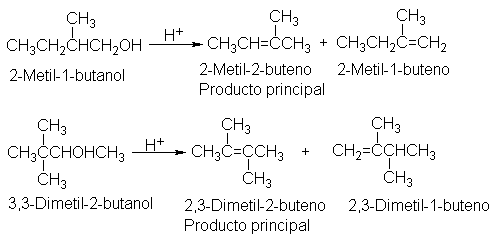
En cada caso, podemos explicar los productos
de la forma usual: el carbocatión formado inicialmente se transpone a otro más
estable. Los alquenos obtenidos son aquéllos que se generan por pérdida de un
protón por este carbocatión transpuesto y por el ion original.
La
orientación es fuertemente Saytzeff.
Donde puede gemnerarse más de un
alqueno, el producto preferido es el más estable. Por ejemplo:
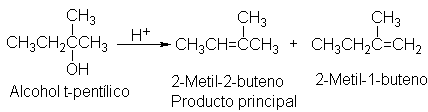
(Obsérvense otra vez los ejemplos de
transposición dados antes.) Desde luego, esto es lo que cabe esperar de la
pérdida de un protón por un carbocatión, como vimos de la sección 7.22.
Interviene aquí otro nuevo factor. Puesto que
la deshidratación es reversible, la composición del producto no refleja
necesariamente qué alqueno se forma más rápidamente, sino, dependiendo de lo
mucho que la reacicón se acerca, al equilibrio, qué alqueno es más estable.
Como sabemos, sin embargo, el alqueno más estable es el generalmente se forma
más rápido. En cualquier caso, la orientación está de acuerdo con el mecanismo,
y las predicicones que hacemos en cuanto a
la orientación, con toda probabilidad son buenas.
Hemos mencionado que los alcoholes
secundarios y terciarios reaccionan por este mecanismo carbocatiónico. Los
alcoholes primarios presentan un problema especial, Vimos (Sec. 5.22) que los
carbocationes primarios se forman
difícilmente. No obstante, la deshidratación de alcoholes primarios produce
transposiciones, tan características de las reacciones carbocatiónicas. Por
ejemplo:
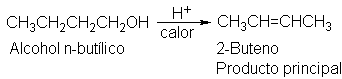
Hay varias explicaciones posibles para lo
anterior. Puede ser que, en el ácido concentrado empleado para deshidratar
alcoholes primarios, se genere un
catión primario fuertemente obstruido, pero capaz de transponerse. Puede ser
que para estos sustratos la deshidratación sea una reación E2 del alcohol
protonado, en cuyo caso, lso alquenos transpuestos no resultan de la
transposición de un catión primario, sino de la reversibilidad de la deshidratación.
(Véase Problema 8.6, Sec. 8.12.)
En la deshidratación observamos otra vez el
papel vital que desempeña la protonación del grupo OH: la transformación de un
grupo saliente pésimo en otro excelente. En la reacción de alcoholes con
halogenuros de hidrógeno (Sec. 5.25), esta transformación posibilita la
sustitución nucleofílica; aquí, permite la eliminación.
En la deshidratación, el alcohol protonado
reacciona, en la mayoría de los casos, por la vía carbocatiónica, como en E1;
por otra parte, los halogenuros de alquilo generalmente siguen E2. Encontramos
la misma situación en la sustituciónnucleofílica (Sec. 5.25), y la explicación
aquí es esencialmente la misma. Para deshidratarse, un alcohol debe protonarse,
por lo que se requiere un medio ácido. En la eliminación E2 necesitamos una
base bastante fuerte para atacar el sustrato sin tener que esperar que éste se
disocie a carbocationes. Pero una base fuerte y un medio ácido son, por
supuesto, incompatibles: toda base más fuerte que el propio alcohol se
protonaría a expensas del alcohol. Por tanto, la deshidratación sigue
generalmente la vía de los carbocationes forzada a tener lugar en ausencia de
una base fuerte. Puesto que los alcoholes son los precursores usuales de los
halogenuros y sulfonatos de alquilo, todas las eliminaciones de este capítulo
son, en cierto sentido, ilustraciones de un mismo punto: la transformación del
OH en un grupo saliente mejor, que se logra con una conversión a halogenuro o
sulfonato de alquilo. Los mismo ocurre con la protonación; es simple, pero
tiene su valor; estamos limitados en nuestra elección del reactivo clave: la
base.