Sustitución electrofílica aromática
(Ir a página principal http://organica1.org/ )
Sustitución electrofílica aromática
2 Efectos de grupos sustituyentes
3 Determinación de la orientación
4.- Determinación de la
reactividad relativa
5.- Clasificación de grupos sustituyentes
6.- Orientación en bencenos disustituidos
8.- Mecansimo de la sulfonación
9.- Mecanismo de la alquilación de Friedel-Crafts
9.- Mecanismo de la halogenación
10.- Desulfonación. Mecanismo de la protonación
11.- Mecanismo de la
sustitución
12.- Mecanismo de la sustitución
14ª.- Teoría de la orientación
15.- Liberación de electrones
por resonancia
16.- Efecto del halógeno en la sustitución electrofílica aromática
1 Introducción
Hemos visto que las relaciones características del benceno implican sustitución, en la que se conserva el sistema anular estabilizado por resonancia. ¿Qué tipos de reactivos permiten esta sustitución? ¿Cuál es el mecanismo de estas sustituciones?
Encima y debajo del plano del
anillo bencénico tenemos una nube electrónica n (Figura 1). Debido a la
resonancia, estos electrones n están más involucrados en mantener núcleos de
carbono unidos que los electrones p de un doble enlace carbono-carbono. De todas formas, y en comparación
con los electrones o, estos electrones p están relativamente sueltos y disponibles
para un reactivo que busca electrones.
No es de extrañar que en
sus reacciones típicas el anillo bencénico sirva de fuente de electrones, esto
es, que actúe como base. Los compuestos con los que reacciona son
electrónicamente deficientes; es decir,
son reactivos electrofílicos o ácidos. Al igual que las reacciones
típicas de los alquenos son de adición electrofílica, las del anillo bencénico
son de sustitución electrofílica.
Estas reacciones no sólo
son típicas del benceno mismo, sino también del anillo bencénico donde quiera
que se encuentre y, de hecho, de muchos
anillos aromáticos, bencenoides y no bencenoides.
La sustitución
electrofílica aromática incluye una amplia gama de reacciones: nitración,
halogenación, sulfonación y reacciones de Friedel-Crafts, experimentadas por
casi todos los anillos aromáticos; procesos como nitrosación y acoplamiento
diazoico, que sólo sufren los anillos de gran reactividad, y reacciones como la
desulfonación, intercambio isotópico y muchos cierres de anillos. Aunque estos aparentemente no
tienen relación alguna, es provechoso
considerarlos como procesos de este tipo, cuando se someten a un examen más
profundo. Desde el punto de vista de su importancia en síntesis, la sustitución
electrofílica aromática quizá no ha sido igualada por ninguna otra clase de
reacciones orgánicas. Constituye la vía de acceso inicial para casi todos los
compuestos aromáticos, pues permite la introducción directa de ciertos grupos
sustituyentes que luego pueden convertirse en otros, incluyendo anillos
aromáticos adicionales, por reemplazo, o por transformación.
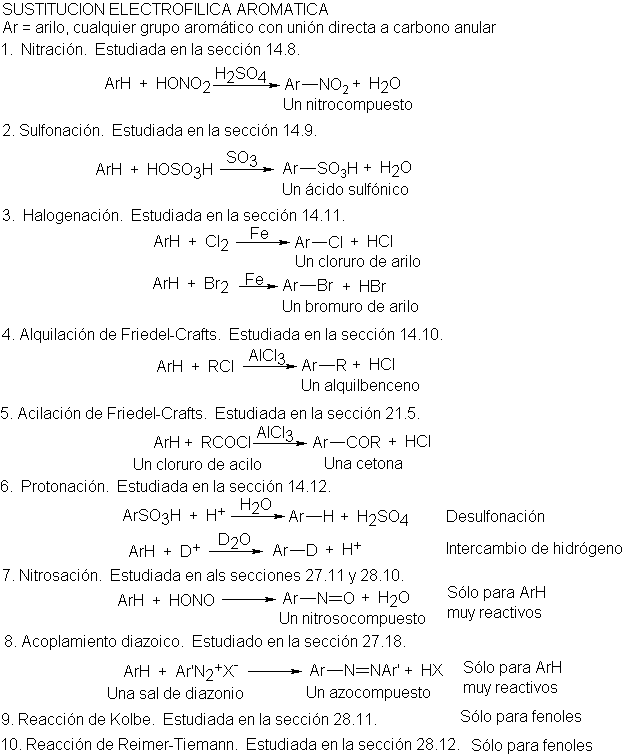
2 Efectos de grupos sustituyentes
Como el benceno, el tolueno
experimenta sustitución electrofílica aromática: sulfonación, por ejemplo,
Aunque son posibles tres productos monosulfonados, en realidad esta reacción
sólo da cantidades apreciables de dos de ellos: los isómeros orto y para.

El benceno y el tolueno son
insolubles en ácido sulfúrico, mientras que los ácidos sulfónicos son muy
solubles. La desaparición de la capa de hidrocarburo indica que se ha completado
la reacción. Al ser agitado a temperatura ambiente con ácido sulfúrico fumante,
el benceno reacciona completamente entre 20 y 30 minutos, mientras que el
tolueno lo hace entre uno y dos minutos.
El estudio de la nitración,
halogenación y alquilación de Friedel-Crafts da resultados análogos. De alguna
manera, el grupo alquilo hace más reactivo al anillo bencénico y que el benceno
no sustituido, y dirige el reactivo
ataque a las posiciones anulares orto y para.
Por otra parte, y para
considerar un ejemplo diferente, se ha encontrado que el nitrobenceno se
sutituye de forma más lenta que el benceno, y que produce principalmente el
isómero meta.
Como el metilo o el nitro,
cualquier grupo unido a un anillo
bencénico lo afecta en su reactividad y determina la orientación de la
sustitución. Cuando un reactivo electrófilo ataca un anillo aromático, el
grupo ya enlazado determina lo fácil
que será el ataque, y dónde sucederá.
Cuando un grupo hace que un
anillo sea más reactivo que el benceno, se llama grupo activante; si produce el
resultado contrario, se conoce como
grupo desactivante.
Un grupo que motiva un
ataque en las posiciones orto y para es un director orto-para; uno que ocasiona
lo mismo en las posiciones meta, se denomina director meta.
En este capítulo
examinaremos los métodos que se emplean para medir estos efectos sobre la
reactividad y la orientación, sus resultados y una teoría que justifica dichos
resultados. Evidentemente, la teoría se basa en el mecanismo más probable para
la sustitución electrofílica aromática. Veremos cuál es este mecanismo y,
también, algunas pruebas que lo apoyan. Primero examinemos los hechos.
3 Determinación de la orientación
En principio, la
determinación del efecto de un grupo sobre la orientación es bastante simple.
El benceno que contiene este grupo se somete a sustitución, y luego se
determina la proproción de los tres isómeros en el producto. Por lo general, la
identificación de cada isómero como orto, meta o para implica compararlo con una muestra auténtica
preparada por otro método, desde una sustancia de estructura conocida. Todas
estas identificaciones se remontan a determinaciones absolutas del tipo Corner
.
De esta manera, se ha
encontrado que cada grupo puede colocarse en una de dos categorías: directores
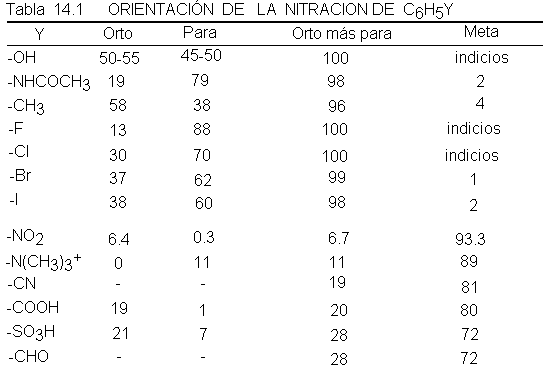
orto-para y directores meta. La tabla
14.1 resume la orientación de la nitración en varios bencenos sustituidos. De
las cinco posiciones abiertas al ataque, tres (60 %) son orto y para con
respecto al sustituyente, y dos (40%), meta; si en la reacción de sustitución
no hubiera selectividad, anticiparíamos que los isómeros orto y para
conformarían el 60% del producto, y el metal, el 40%. En cambio, observamos que
siete de los grupos dirigen el 96-100% de la nitración a las posiciones orto y
para; los seis restantes dirigen el 72-94% a las posiciones meta.

Un grupo determinado
ocasiona el mismo general de orientación predominantemente, orto, para, o
principalmente, meta cualquiera que sea el reactivo electrofílico implicado.
Sin embargo, la distribución efectiva de los isómeros pueden variar de una
reacción a otra. Por ejemplo, en la
tabla 14.2 se compara la distribución de isómeros obtenida del tolueno por
sulfonación o bromación con la lograda por nitración.
4.- Determinación de la reactividad relativa
Un grupo se clasifica como
activante si el anillo al cual sustituye es más reactivo que el benceno, y
desactivante, si sucede lo contrario. Las reactividades del benceno y de uno
sustituido se comparan de una de las maneras siguientes.
Puede medirse el tiempo que
requieren las reacciones en condiciones idénticas. Así, vimos que el tolueno
reacciona con ácido sulfúrico fumante entre una décima y una vigésima parte del
tiempo requerido por el benceno: el tolueno es más reactivo que el benceno, por
lo que CH3 es un grupo activante.
Puede observarse la severidad de las condiciones
requeridas para que se verifique una reacción comparable dentro del mismo periodo
de tiempo. Así, por ejemplo, el benceno se
nitra en menos de una hora a 60ºC con una mezcla de ácidos nítrico y
sulfúrico concentrados. Una nitración comparable del nitrobenceno requiere un
tratamiento a 90ºC con ácido nítrico fumante y ácido sulfúrico concentrado.
Evidentemente, el nitrobenceno es menos
reactivo que el benceno, y el grupo nítro, NO2, es
desactivante.
Para una comparación
cuantitativa exacta en condiciones idénticas de reacción, pueden realizarse
reacciones competitivas, donde los compuestos a comparar compiten por una cantidad
limitada de un reactivo. Si, por ejemplo,
se mezclan cantidades equimolares de
benceno y tolueno con una cantidad pequeña de ácido nítrico en un disolvente se
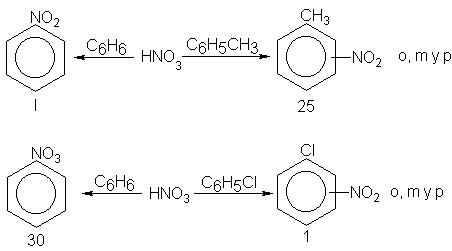
obtinene como alrededor de 25 veces más nitrotolueno que nitrobenceno,
indicando que el tolueno es 25 veces más reactivo que el benceno. Por otra
parte, una mezcla de benceno y clorobenceno da un producto en el que el
nitrobenceno es sólo un trigésimo de la del benceno. Por tanto, el grupo cloro
se clasifica como desactivante, y el metilo, como activante. La activación o
desactivación ocasionada por algunos grupos es extremadamente poderosa: la
anilina, C6H5NH2, es casi un millón de veces
más reactiva que el benceno, y el nitrobenceno, C6H5NO2,
tiene alrededor de un millonésimo de la reactividad del benceno.
5.- Clasificación de grupos sustituyentes
Los métodos descritos en
las dos últimas secciones han sido utilizados para determinar los efectos de gran
número de grupos sobre la sustitución electrofílica. Como se ilustra en la
tabla 14.3, casi todos los grupos caen en una de dos clases: activantes y
directores orto-para, o desactivantes y directores meta. Los halógenos forman
una clase aparte, pues son desactivantes, pero directores orto y para.

Con sólo conocer los
efectos que se resumen en esta corta lista, podemos predecir con bastante
exactitud el curso de cientos de reacciones de sustitución aromática. Por ejemplo,
sabemos ahora que la bromación del nitrobenceno dará principalmente el isómero
meta, y que la reacción procederá más lentamente que la bromación del benceno. Probablemente,
se requieran condiciones severas para
que llegue a realizarse. Sabemos ahora, por ejemplo, que la nitración de
C6H5NHCOCH3 (acetanilida) producirá esencialmente los isómeros orto y para, y
que será más rápida que la nitración del benceno.
Como veremos, a pesar de
poder explicar estos efectos racionalmente, es necesario que el estudiante
memorice las clasificaciones de la tabla
14.3, para que pueda tratar los problemas de síntesis de compuestos aromáticos con rapidez.
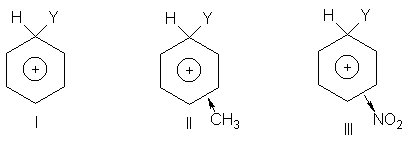
6.- Orientación en bencenos disustituidos
La presencia de dos
sustituyentes en un anillo complica el problema de la orientación, pero aun así
frecuentemente pueden hacerse pronósticos muy definidos. En primer lugar, ambos
sustituyentes pueden estar ubicados de modo tal, que la influencia directora de
uno refuerce la del otro; por ejemplo, en I, II y III, la orientación debe ser
claramente la indicada por las flechas.

Por otra parte, cuando el
efecto director de un grupo es opuesto
al del otro, puede ser difícil predecir el producto principal. En tales casos,
a menudo se obtienen mezclas complejas de varios compuestos.
Sin embargo, aun cuando hay
efectos opuestos, es posible hacer predicciones de acuerdo con las generalizaciones siguientes.
(a) Los grupos activantes poderosos por lo general
se imponen a los desactivantes o activantes débiles. Las diferencias en el poder
director en la serie
![]()
Son suficientemente grandes
como para emplearlas en la planificación de síntesis factibles. Por ejemplo:
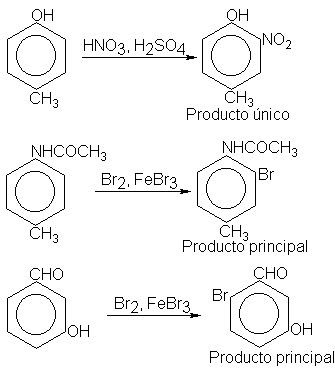
Sin embargo, debe haber una
diferencia suficientemente grande entre los efectos de ambos grupos para
obtener resultados claros; de otro modo, puede obtenerse un resultado como el
siguiente:
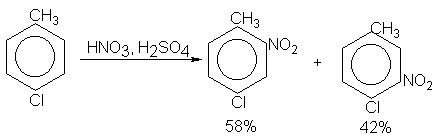
(b) A menudo se produce poca sustitución entre dos grupos que sean
recíprocamente meta. En muchos casos, da la impresión de que no hay suficiente
espacio entre dos grupos en posiciones meta entre sí como para que haya
sustituido apreciable, según se ilustra en IV y V:
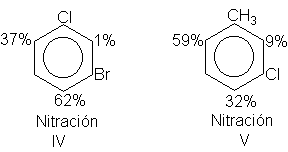
7.- Orientación y síntesis
Como se dijo anteriormente,
una síntesis de laboratorio tiene por finalidad la obtención de un solo
compuesto puro. De ser posible, debemos evitar el uso de reacciones que
produzcan mezcla, puesto que disminuye el rendimiento de la sustancia deseada y
causa problemas difíciles de purificación. Después de tener esto en mente,
veamos algunas posibildiades de aplicación de nuestros conocimientos sobre la
orientación en la síntesis de compuestos aromáticos puros.
En primer lugar,
consideremos el orden en que introducimos los diversos sustituyentes en el
anillo. Así, en la preparación de los bromonitrobencenos es evidente que si
nitramos primero y luego bromamos, obtenemos el isómero meta; en cambio, si
bromamos primero y después nitramos, obtenemos una mezcla de los isómeros orto
y para. Por consiguiente, el orden utilizado dependerá del isómero que deseamos.
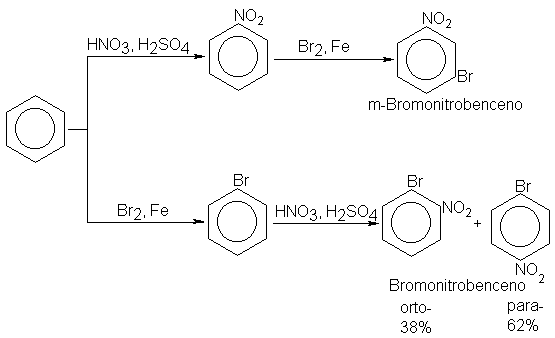
Si, a continuación, nuestra
síntesis implica la conversión de un grupo en otro, consideraremos el momento oportuno
para esta conversión. La oxidación de un grupo metilo, por ejemplo, da un carboxilo. En la preparación
de los ácidos nitrobenzoicos desde el tolueno, el producto específico obtenido
depende de que se oxide o nitre primero.
La sustitución controlada
por un activante da una mezcla de los isómeros orto y para.
Sin embargo, a menudo
debemos hacer uso de dichas reacciones, como en los ejemplos presentados. Por
lo general, es posible obtener el isómero para puro por cristalización fraccionada de
la mezcla, que al ser el isómero el más simétrico es el menos soluble, y
cristaliza mientras el disolvente aún retiene el soluble isómero orto. Por
supuesto, algo del isómero para permanece en la solución, contaminando al
isómero orto, que es, por tanto, difícil de purificar. Como veremos, a menudo
se usan procedimientos especiales para preparar isómeros orto.
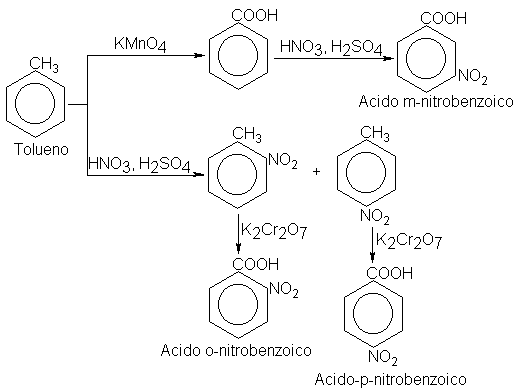
En el caso especial de los
nitrocompuestos, las diferencias en sus puntos de ebullición son suficientemente apreciables como para poder
obtener los isómeros orto y para en forma pura por destilación fraccionada.
Como resultado, muchos compuestos aromáticos se preparan mejor no por
sustitución directa, sino por conversión de
un grupo en otro. A fin de cuentas, se parte originalmente de un
nitrocompuesto. Estos métodos de conversión los estudiaremos más adelante.
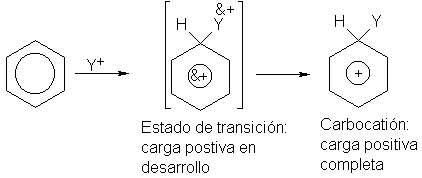
8.- Mecanismo de la nitración
Ahora que hemos apreciado
los efectos que ejercen los grupos sustituyentes sobre la orientación y la
reactividad en la sustitución electrofílica aromática, veamos si podemos justificarlos. El primer paso es examinar el
mecanismo para la reacción. Comencemos con la nitración utilizando benceno como
sustrato aromático.
El mecanismo generalmente
aceptado para la nitración con una mezcla de ácidos sulfúrico y nítrico (la
mezcla ácida tan empleada por los
químicos orgánicos) implica la serie de reacciones siguientes:
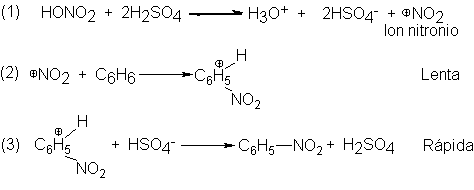
El paso (1) genera el ión
nitronio, NO2+,
que es partícula electrófila que realmente ataca al anillo bencénico. Esta reacción es un equilibrio ácido-base,
en el que el ácido sulfúrico sirve de ácido, y el ácido nítrico, mucho más
débil, de base. Podemos considerar que el ácido hacerlo en el usual H+...-ONO2. El ión nitronio es bien
conocido, puesto que existe en sales como el perclorato de nitronio, NO2 + CIO4-, y el fluoroborato de
nitronio, NO2+ BF4-. De hecho,
George Olah comprobó que estas sales de
nitronio estables, disueltas en disolventes como el nitrometano o el ácido
acético, nitran compuestos aromáticos suavemente, a temperatura ambiente y con
rendimientos elevados.
Como necesita electrones,
el ión nitronio los encuentra particularmente disponibles en la nube n del
anillo bencénico, de modo que en el paso (2) se une a uno de los carbonos por
medio de un enlace covalente, generando el carbocatión
Que a menudo se denomina ión
bencenonio.
¿Cuál es la estructura de
este carbocatión? Podemos representarlo por tres estructuras (I, II y III), que
sólo difieren en las ubicaciones de los dobles enlaces y de la carga positiva.
Por tanto, el verdadero ión debe ser un híbrido de resonancia de estas tres
estructuras.
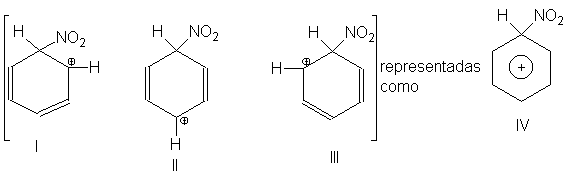
Desde luego, esto significa
que la carga positiva no se encuentra localizada sobre un sólo átomo de
carbono, sino que está distribuida sobre la molécula, y es particularmente
intensa sobre los carbonos orto y para, con respecto al que lleva el grupo NO2.
(Más adelante veremos que esta distribución orto-para es significativa.) La dispersión
de la carga positiva sobre la molécula
por la resonancia estabiliza este ión con respectoa uno con carga localizada.
Considerando la estabilidad del benceno original, que este ión llegue a
formarse probablemente se debe a esa estabilización por resonancia. A veces, el
carbocatión híbrido se representa como en IV, donde la línea de puntos
representa enlaces parciales debido a los electrones n deslocalizados.
Hasta aquí, la reacción es
similar a la adición a alquenos: una
partícula electrófila, atraída por los
electrones n, se une a la molécula para generar un carbocatión. Pero el destino
de este ión es diferente al del correspondiente a un alqueno; la unión de un
grupo básico al ión bencenonio para dar el producto de adición destruiría el
carácter aromático del anillo. En cambio, el ión básico, HSO4-, quita un ión
hidrógeno (paso 3) para dar el producto de sustitución, que retiene el anillo
estabilizado por resonancia. Habíamos visto que la pérdida de un protón es una
de las reacciones típicas de un
carbocatión . En este caso, es la reacción preferencial.
Como en otras reacciones de
carbocationes analizadas, el paso más
difícil es la formación del carbocatión (paso 2). Una vez generado, el
carbocatión pierde rápidamente un ión hidrógeno (paso 3) para dar los
productos. (Daremos pruebas de esto en Sec. 14.)
En consecuencia, la sustitución
electrofílica es un proceso por etapas, igual que la adición electrofílica, que
pasa por un carbocatión intermediario. Sin embargo, los dos procesos difieren
en el destino del carbocatión. Aunque el mecanismo de la nitración pueda ser el
mejor establecido entre los mecanismos de las demás reacciones de sustitución aromática, resulta claro que
todas las reacciones siguen el mismo curso.
8.- Mecansimo de la sulfonación
La sulfonación de muchos
compuestos aromáticos implica los pasos siguientes:
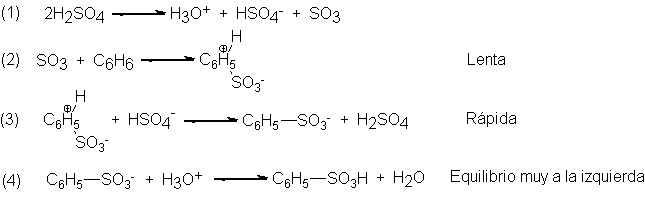
De nuevo, el primer paso,
que genera el trióxido de azufre electrófilo, es simplemente un equilibrio ácido-base, esta vez entre
moléculas de ácido sulfúrico. Para la sulfonación utilizamos a menudo ácido
sulfúrico con un exceso de SO3. Aunque cuando no se haga esto, el
electrófilo es SO3, formado en el
paso (1).
![]()
En el paso (2), el reactivo electrófilo, SO3,
se une al anillo bencénico para generar el carbocatión intermediario. Aunque el
trióxido de azufre no tiene carga positiva, es electrónicamente deficiente y,
por tanto, ácido.
El paso (3) es la pérdida de un protón para dar el producto de sustitución estabilizado por resonancia: esta vez el anión del ácido bencenosulfónico se disocia fuertemente, por ser un ácido fuerte (paso 4).
¨Con algunos sustratos aromáticos y a cierta acidez, el
electrófilo puede se HSO3+ o molécula que transfiere con
afcilidad SO3 o HSO3+ al anillo aromático.
9.- Mecanismo de la alquilación de Friedel-Crafts
En la alquilación de
Friedel-Crafts, el electrófilo típico es un carbocatión generado por un
equilibrio ácido-base, esta vez en el sentido de Lewis:
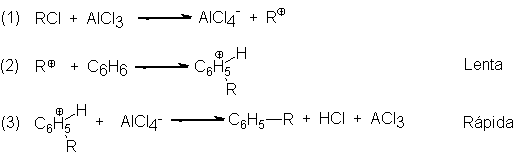
En ciertos casos, no se
halla involucrado un carbocatión libre. En cambio, se transfiere el grupo alquilo con un par de electrones menos
directamente al anillo aromático desde el complejo polar, I, que forman el AICI3
y el halogenuro de alquilo:
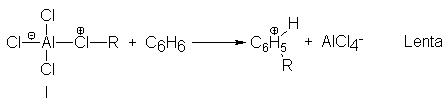
El electrófilo es entonces
(a) R+ o (b) una molécula como I, que puede transferir con facilidad
R+ al anillo aromático. Esta dualidad de mecanismo es corriente en la sustitución
electrofílicaa aromática. En ambos casos, el ácido de Lewis R+ es desplazado de RCI por el otro ácido de
Lewis, el AICI3.
Describimos la reacción de Friedel-Crafts como una sustitución electrofílica y, desde el punto de vista del anillo aromático, efectivamente lo es. Sin embargo, como un ácido reacciona con una base, también un electrófilo reacciona con un nucleófilo, una molécula que proporciona los electrones que busca el primero. Por consiguiente, desde el punto de vista opuesto, esta reacción consiste en un ataque nucleofílico del anillo aromático sobre el grupo del complejo I. El ión AICI4- es un grupo saliente mejor que el CI-; aquí, el ácido de Lewis, AICI3, cumple el mismo propósito que un ácido de Lowry-Bronsted en la protonación de un alcohol.
Cuando consideremos la
reacción de Friedel-Crafts como herramienta de síntesis, descubriremos que, en
su sentido más amplio, esta reacción implica reactivos diferentes de los
halogenuros de alquilo, y otros ácidos de Lewis que el cloruro de aluminio: BF3,
SnCI4, HF, aun H+.
9.- Mecanismo de la halogenación
La halogenación aromática
implica los pasos que se ilustran para la cloración.

El paso clave (2) es la
unión de cloro positivo al
anilloaromático. Sin embargo,
parece poco probable que esté involucrado un ión CI+ libre. En
cambio, se combina cloruro
férrico con CI2 para
formar el complejo II, del que
se transfiere cloro, sin sus electrones, directamente al anillo.
Hemos visto que la adición
de halógenos a los alquenos consiste
también en un ataque con halógeno positivo para formar un carbocatión
intermediario. Sin embargo, los electrones p muy sueltos del alqueno lo hacen más
reactivo, por lo que se transfiere halógeno positivo directamente de la molécula
X2, con pérdida de CI -. La molécula de benceno menos reactiva necesita la ayuda de un a´cido de Lewis, y la
reacción procede con la pérdida del mejor grupo saliente, el FeCI4-. Sin embargo, los compuestos aromáticos más reactivos, es decir,
aquellos con electrones n más disponibles, reaccionan con halógenos en ausencia de ácidos de Lewis.
10.- Desulfonación. Mecanismo de la protonación
Cuando se calienta un ácido sulfónico aromático con
ácido acuoso hasta 100-175º, se convierte en ácido sulfúrico y un hidrocarburo
aromático. Esta desulfonación es la inversión exacta del proceso de sulfonación
por el que se obtuvo el ácido sulfónico original.
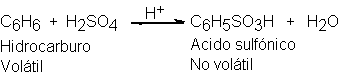
Aplicando los principios
usuales de los usuales de los
equilibrios, podemos elegir
condiciones que lleven la reacción en la dirección que nos convenga. Para
sulfonar, empleamos un gran exceso de ácido sulfúrico concentrado o fumante: la
alta concentración del agente sulfonante y una baja concentración de agua ( o
su eliminación por reacción con SO3) desplaza el equilibrio hacia la
formación del ácido sulfónico. Para
desulfonar, usamos ácido diluido y a menudo pasamos vapor sobrecalentado por la
mezcla reaccionante: la elevada
concentración de agua y la eliminación del relativamente volátil hidrocarburo
por destilación con vapor, desplaza el equilibrio hacia el hidrocarburo.
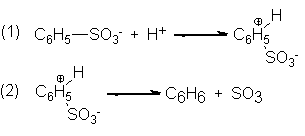
De acuerdo con el principio de reversibilidad
microscópica, el mecanismo de desulfonación debe corresponder
exactamemnte a la inversión del de sulfonación. Esta reacción es otro ejemplo
de sustitución electrofílica
aromática: el electrófilo es el protón, H+, y la reacción es
una protonación o,
más específicamente, una protodesulfonación.
La sulfonación es excepcional entre las reacciones
de sustitución electrofílica aromática. También lo
es en otro aspecto: en la
sulfonación, el hidrógeno
ordinario (protio) es desplazado
de un anillo aromático
unas dos veces más rápidamente
que el deuterio. Ambas propiedades
están relacionadas y dan,
como apreciaremos en la sección 14, una
descripción más detallada
de la sulfonación y de la sustitución
electrofílica aromática en
general.
11.- Mecanismo de la sustitución
Electrofílica aromática:
resumen
Cualquiera que sea el
reactivo específico involucrado, parece que las reacciones de sustitución
electrofílica aromática proceden por un solo mecanismo. Para el reactivo YZ esto puede resumirse como sigue:
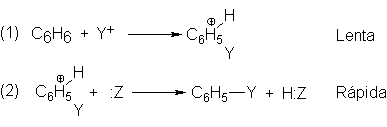
El mecanismo comprende dos pasos esenciales: (1) el ataque de un reactivo electrofílico
al
Anillo, para formar un
carbocatión,
Y (2) extracción de un
protón al carbocatión por alguna base.
En cada caso hay una reacción ácido-base preliminar que genera la
partícula atacante. Sin embargo, la verdadera sustitución está contenida en
estos dos pasos.
La mayor parte del apoyo a
este mecanismo proviene de pruebas sobre la naturaleza de la partícula ataque
en cada una de estas reacciones; es decir, evidencia del carácter electrofílico
de la sustitución. A su vez, estas pruebas provienen en gran medida de estudios
cinéticos, complementados con otras observaciones parecidas a las de
carbocationes en algunas alquilaciones de Friedel-Crafts . La naturaleza
electrófila de estas reacciones se ve apoyada ampliamente por el hecho de que
otros procesos que muestran las mismas características de reactividad y
orientación también se ajustan al mismo esuqema del mecanismo.
Veamos ahora pruebas de
otro tipo: la evidencia de efectos isotópicos.
12.- Mecanismo de la sustitución
Electrofílica aromática: dos etapas
Hasta ahora sólo hemos
hablado de pruebas que indican que estas reacciones son electrofílicas, y
revelan cuáles son los electrófilos más probables. Sin embargo, lo anterior
representa sólo parte del mecanismo. Aceptando que la sustitución es electrofíla,
¿cómo sabemos que la sustitución electrofílica aromática implica dos etapas,
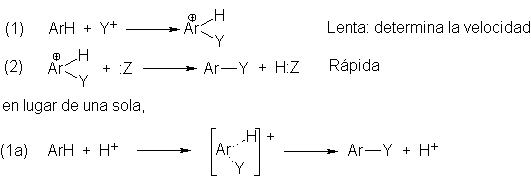
¿Cómo sabemos que, de estas
dos etapas, la primera es mucho más lenta que la segunda?
La respuesta se encuentra
en una serie de estudios iniciados por Lars
Melander (Universidad de Götenburgo) y ampliados por muchos otros
investigadores. Se sometieon a nitración, bromación y alquilación de
Friedel-Crafts varios compuestos aromáticos marcados con deuterio o tritio, y
se encontró que en todas estas reacciones el deuterio o el tritio es
reemplazado a la misma velocidad que el protio: no hay efecto isotópico
significativo.
Hemos visto que un enlace carbono-deuterio se rompe más
lentamente que un carbono-protio, y un enlace carbono-protio, y un enlace
carbono-tritio, aún más lentamente. Tales efectos isotópicos primarios son
considerables: KH/KD puede valer de 5 a 8, y KH/KT, alrededor del doble. Luego,
¿cómo interpretamos el hecho de que no hay efecto isotópico en estos casos? Si la velocidad del reemplazo de los
diversos isótopos del hidrógeno es la misma, sólo puede significar que las
reacciones cuya velocidad estamos comparando no implican la ruptura de un
enlace carbono-hidrógeno.
Esta interpretación
concuerda con nuestro mecanismo. La velocidad de la sustitución completa está
determinada por la unión lenta del reactivo electrofílico al anillo aromático
para formar el carbocatión, que, una vez generado, pierde rápidamente un ión hidrógeno protón o deuterón para dar el
producto. Por consiguiente, la etapa (1) es la que determina la velocidad.
Puesto que no implica la ruptura de un enlace carbono-hidrógeno, su velocidad
y, en consecuencia, la de todo el proceso es independiente del isótopo
específico de hidrógeno presente.
Si la sustitución consistiera
en una sola etapa, como en (1ª), ésta necesariamente determinaría la velocidad y, puesto que comprende la ruptura de
un enlace carbono-hidrógeno, se observaría un efecto isotópico. O bien, si la
etapa (2) de la serie de dos pasos fuera suficientemente lenta con respecto a
la (1) en afectar a la velocidad global, también observaríamos un efecto
isótopico. La sulfonación muestra, de hecho, un pequeño efecto isótopico y,
como se verá más adelante, precisamente por esta razón. Sin embargo, incluso en
la sulfonación, el control de la velocidad total lo ejerce la etapa (1).
Por tanto, la ausencia de
efectos isótopicos establece no sólo las dos etapas de la sustitución
electrofílica aromática, sino también la velocidad relativa de cada una. La
unión del electrófilo a un átomo de carbono del anillo es el pasos difícil
(véase Fig. 14.2), pero sería
igualmente difícil si el carbono llevara protio o deuterio. El paso siguiente,
la pérdida de un ión hidrógeno, es fácil. Aunque más lenta para el deuterio que
para el protio, en realidad no implica diferencia: ligeramente más lenta para
el deuterio que para el protio, en relaidad no implica diferencia: ligeramente
más rápida o más lenta, su velocidad no afecta a la del proceso completo.
Consideremos este punto más
en detalle (Fig.14.2, detalle). Cada carbocatión formado, I(H) o I(D), continúa
a producto, puesto que la barrera energética hacia la derecha (por delante de
carbocatión)-ligeramente más alta para el deuterio o

ligeramente más baja para
el protio es considerablemente inferior que hacia la izquierda (detrás del
carbocatión); pero esta última es Eact para la etapa (1) invertida. La reacción
inversa debe ser mucho más lenta que la etapa (2), si la (1) verdaderamente ha
de ser la que determine la velocidad . Resumiendo en función de las constantes
de velocidad, K, para las diversas etapas, tenemos:
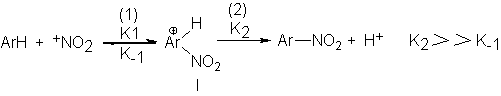
Podemos apreciar por qué la
nitración y reacciones similares no son reversibles. En la inversa de la
nitración, el nitrobenceno (la reacción 2 invertida) se protona para formar el
carbocatión I; por supuesto, éste es el mismo que el ión I generado en la
nitración, y hace lo mismo: (re) genera nitrobenceno.
A diferencia de la mayoría
de las otras reacciones de sustitución, la sulfonación presenta un efecto
isotópico moderado: el hidrógeno ordinario (protio) es desplazado de un anillo
aromático casi el doble de rápido que el deuterio.¿Significa esto que la
sulfonación procede por un mecanismo diferente que la nitración, uno que conste
de una sola etapa? Es casi indudable que no.
La sulfonación se distingue
de la mayoría de las reacciones de sustitución electrofílica por ser
reversible. Esto nos da la pista. La reversibilidad significa que el
carbocatión II puede perder SO3 para fromar
el hidrocarburo. Evidentemente, en este caso la reacción (2) no es mucho más
rápida que la reacción invertida (1). En la sulfonación, las barreras
energéticas a ambos lados del carbocatión II deben ser aproximadamente de la
misma altura: algunos iones siguen en una dirección y otros en la
contraria (Fig. 14.3). Ahora bien, tanto si el carbocatión es II(D) como si es
II(H), la barrera izquierda (detrás de él) es de la misma altura. Pero para
subir la barrera derecha (por delante) debe romperse un enlace
carbono-hidrógeno, por lo que esta barrera es más elevada para el carbocatión
II(D) que para el II(H). Vuelve más iones deuterados que ordinarios al material
inicial, de modo que la sulfonación total es más lenta para el benceno
deuterado. Por tanto, la forma particular de la curva de energía potencial que
permite la reversibilidad de la sulfonación, también admite observar un efecto
isotópico.
Utilizando sustratos
aromáticos seleccionados especialmente los muy impedidos pueden detectarse
efectos isotópicos en otros tipos de sustitución aromática, incluso en la
nitración. En ciertas reacciones pueden variarse deliberadamente el tamaño del
isótopo por medio de cambios en las condiciones experimentales, de forma que
aparece una dependencia de la velocidad relativa de (2) y la inversa de (1). No
hay duda de que todas estas reacciones siguen el mismo mecanismo de dos etapas,
pero con diferencia en als formas de las curvas de energía potencial. Con los efectos isotópicos, el químico
dispone de una sonda muy delicada para el estudio de los mecanismos de
reacciones orgánicas.
13.-Reactividad y orientación
Hemos visto que
ciertos grupos activan el anillo
bencénico y dirigen la sustitución a las posiciones orto y para, mientras otros
lo desactivan y dirigen la sustitución a posiciones meta (excepto los
halógenos). Veamos si pueden justificarse estos efectos de acuerdo con los
principios recién aprendidos.
En primer lugar, debemos recordar que reactividad y
orientación son una cuestión de la velocidad relativa de reacción. Se dice que
el metilo activa el anillo porque lo hace reaccionar más rápidamente que el benceno;
orienta a orto y para porque hace que esas posiciones reaccionen más
rápidamente que las meta.
Sabemos que la velocidad de
la sustitución electrofílica aromática queda determinada por la misma etapa
lenta, cualquiera que sea el reactivo implicado; el ataque del electrófilo
sobre el anillo para generar un carbocatión:
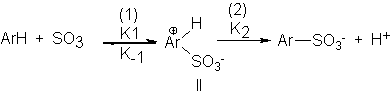
Por tanto, toda diferencia
en la velocidad de la sustitución se debe a diferencias en la velocidad de esta etapa.
Para reacciones muy
relacionadas, una diferencia en la velocidad de formación de carbocationes está
determinada en gran medida por una diferencia en Eact es decir, por
una diferencia en la estabilidad de los estados de transición. Como en otras
reacciones de carbocationes estudiadas, los factores que estabilizan al ión por
dispersión de la carga positiva deben estabilizar, por la misma razón, al
carbocatión incipiente del estado de transición. Aquí también cabe esperar que
el carbocatión más estable se forme con mayor velocidad. Por tanto, nos
encontraremos en las estabilidades relativas de los carbocationes.
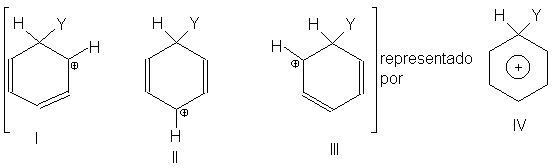
En la sustitución
electrofílica aromática, el carbocatión intermediario es un híbrido de
estructuras I, II y III, donde la carga positiva se distribuye alrededor del
anillo, siendo más fuerte en las posiciones orto y apra con respecto al átomo
de carbono atacado.
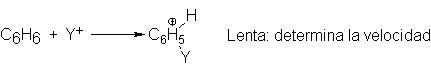
Un grupo ya unido al anillo bencénico debe afectar a la estabilidad
del carbocatión, dispersando o intensificando la carga positiva, según su
naturaleza sea de atracción o liberación de electrones. Por la estructura del ión (I-III), resulta evidente que el efecto
estabilizador o desestabilizador debe ser especialmente importante cuando dicho
grupo tenga una unión orto o para con el átomo de carbono atacado.
14.- Teoría de la reactividad
Para comparar la velocidad
de sustitución en el benceno, tolueno y
nitrobenceno, confrontamos las estructuras de los carbocationes que genera cada
uno de los tres compuestos:
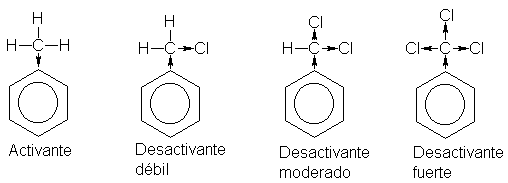
El grupo metilo (II) tiende
a neutralizar la carga positiva liberando electrones y haciéndose, por tanto,
más positivo a sí mismo. Esta dispersión de la carga estabiliza el carbocatión.
El efecto inductivo estabiliza de la misma forma la carga positiva que se
desarrolla en el estado de transición y la reacción se acelera.
En cambio, el grupo –NO2
ejerce un efecto inductivo de atracción de electrones (III): esto tiende
a intensificar la carga positiva, desestabiliza el carbocatión y hace que la
reacción sea más lenta.
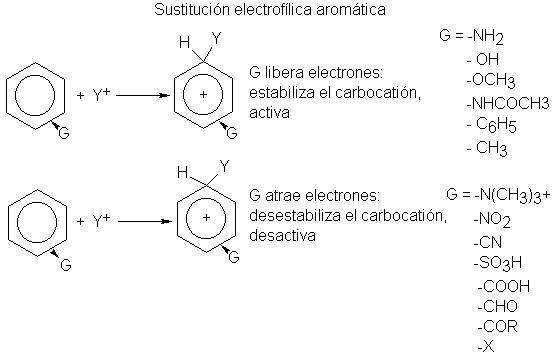
Así pues, la reactividad en la sustitución electrofílica aromática depende de la tendencia de un grupo sustituyente a la liberación o atracción de electrones. Un grupo que libera electrones activa el anillo, y uno que los atrae, lo desactiva.
Otros grupos alquilo liberan electrones, igual que el –CH3
y, como éste, activan el anillo. Por ejemplo, el t-butilbenceno es 16 veces más
reactivo que el benceno en la nitración. La liberación de electrones por NH2
y OH, y por sus derivados OCH3 y NHCOCH3, no se debe a su
efecto inductivo, sino a la resonancia. Esto se estudiará más adelante (Sec.18).
Nos hemos familiarizado ya
con el efecto de atracción electrónica
de los halógenos. La carga positiva
completa del grupo N(CH3)3+ tiene, por supuesto, un fuerte poder de
atracción electrónica. En los otros grupos desactivantes (como NO2, - CN, - COOH), el átomo
adyacente al anillo está unido a oxígeno o nitrógeno por medio de un enlace
múltiple. Estos átomos electronegativos atraen los electrones n móviles,
dejando electrónicamente deficiente al átomo adyacente al anillo. Para
compensar esta deficiencia, dicho átomo extrae electrones del anillo.
Sería de esperar que el
reemplazo de hidrógeno en el -CH3, por
halógeno, redujera la tendencia a liberar electrones del grupo, convirtiéndolo
quizá en otro que los libere, y efectivamente ése es el caso. En la nitración,
el tolueno es 25 veces más reactivo que el benceno; el cloruro de bencilo tiene
un tercio de la reactiviedad del benceno, por lo que el grupo –CH2CI es
desactivante débil. Un reemplazo adicional de hidrógeno por halógeno para dar
los grupos -CHCI2 y –CCI3, resulta en
una desactivación más fuerte.
14ª.- Teoría de la orientación
Antes de justificar la
orientación en la sustitución electrofílica, examinemos los hechos.
Un grupo activante activa
todas las posiciones del anillo bencénico: incluso las meta son más reactivas
que cualquier otra posición individual del propio benceno. Dirige a orto y para
porque las activas más que a la meta.
Un grupo desactivante
desactiva todas las posiciones del anillo, incluso las meta. Dirige meta
simplemente, porque desactiva las posiciones orto y para más que a las meta.
Así, tanto la orientación
orto-para como la meta surgen de la misma forma: el efecto de cualquier grupo
sea activante o desactivante es más intenso en las posiciones orto y para.
Para apreciar si esto es lo
esperado, comparemos, por ejemplo, los carbocationes generados por el ataque en
las posiciones para y meta del tolueno, un compuesto con un grupo activante.
Cada uno de ellos es un híbrido de tres estructuras: I-III para para, IV-VI
para meta. En una de estas seis estructuras, la II, la carga positiva se
encuentra ubicada en el carbono que tiene el –CH3. Aunque este
libera electrones para todas las posiciones del anillo, lo hace más
intensamente hacia el carbono más cercano.
Por consiguiente, la estructura II es una particularmente estable. Debido a la contribución de esta estructura
II, el carbocatión híbrido que resulta del ataque a la posición para es más
estable que el resultante del ataque a la posición meta. Por tanto, la sustitución
es más rápida en para que en meta.
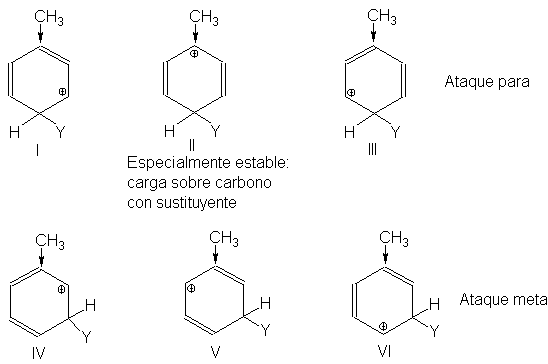
De igual forma, puede
apreciarse que el ataque a una posición orto (VII a IX) también genera un
carbocatión más estable, por la contribución de IX, que el ataque en meta.
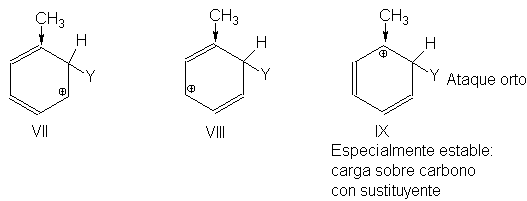
Consecuentemente, en el tolueno,
la sustitución orto-para es más rápida que la meta, porque la liberación de
electrones por -¨CH3 es más efectiva durante el ataque a las
posiciones orto y para con respecto a él.
Comparemos, a continuación,
los carbocationes formados por el ataque a las posiciones para y meta del
nitrobenceno, un compuesto con un grupo desactivante. Cada uno de ellos es un
híbrido de tres estructuras, X a XII para el ataque para, y XIII a XV para el
meta. En una de las seis estructuras, la XI, la carga positiva está ubicada en
el átomo de carbono que porta el grupo
-NO2. Aunque NO2
atrae electrones desde todas las posiciones, lo hace más intensamente desde el
carbono más cercano, por lo que éste, ya positivo, demuestra poca
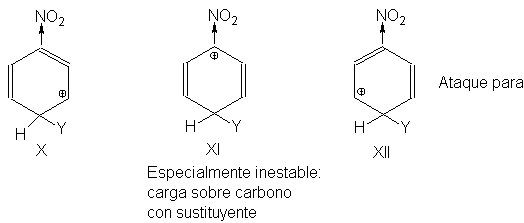
Tendencia a acomodar la
carga positiva del carbocatión. Por tanto, la estructura XI es particularmente
inestable y no contribuye a estabilizar el ión que resulta del ataque en la
posición para. Se extiende que para el ataque en la posición para, el ión es un
híbrido de sólo dos estructuras, X y XII; la carga positiva se restringe
principalmente a sólo dos átomos de carbono. Es menos estable que el ión que
resulta del ataque en la posición meta, que es un híbrido de tres estructuras,
con la carga positiva acomodada por tres átomos de carbono. En consecuencia, la
sustitución para ocurre más lentamente que la meta.
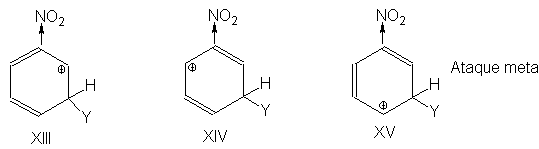
Igualmente, puede
apreciarse que el ataque en la posición orto (XVI a XVIII) genera un
carbocatión menos estable, debido a la inestabilidad de XVIII, que un ataque en
una posición meta.
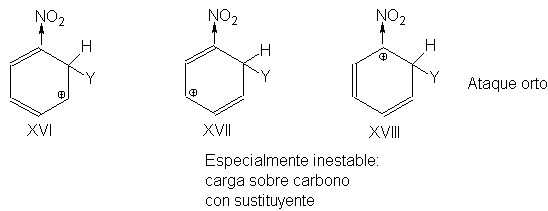
Por tanto, en el
nitrobenceno la sustitución orto, para es más lenta que la meta, porque la
atracción electrónica del NO2 es más efectiva durante el ataque en
las posiciones orto y para con respecto a él.
Vemos así que tanto al orientación orto, para por grupos activantes, como la meta por grupos desactivantes, son consecuencias lógica de la estructura del carbocatión intermediario, cuya carga es máxima en las posiciones orto y para con respecto al punto de ataque Un grupo unido a esas posiciones puede ejercer allí su efecto más intenso, tanto activado como desactivado.
El comportamiento anormal
de los halógenos que dirigen orto y para, a pesar de ser desactivante, resulta de
una combinación de dos factores que se oponen. Esto será estudiado en la
sección 19.
15.- Liberación de electrones por resonancia
En la sustitución
electrofílica aromática, según vimos, un grupo sustituyente afecta a la
reactividad y la orientación por su tendencia a traer o liberar electrones. De
momento, sólo hemos considerado esta tendencia en función de efectos
inductivos; es decir, como efectos debidos a la electronegatividad del grupo
implicado.
Sin embargo, ciertos (-NH2 y –OH, y sus derivados) actúan
como activantes poderosos en la sustitución electrofílica aromática, a pesar de
contener átomos electronegativos, para los cuales puede comprobarse que, en
otras situaciones, ejercen efectos inductivos de atracción de electrones. Si
nuestro enfoque del problema es correcto, estos grupos deben donar electrones
por otro medio que no sean sus efectos inductivos. Se cree que lo hacen por un
efecto de resonancia. Pero antes de analizar esto último, revisemos un poco lo
estudiado sobre el nitrógeno y el oxígeno.
A pesar de ser
electronegativo, el nitrógeno del grupo NH2 es básico y tiende a
compartir su último par de electrones para adquirir una carga positiva. Al
igual que el amoniaco acepta un protón para formar el ión amonio (NH4+),
los compuestos orgánicos relacionados con el amoniaco aceptan protones para
iones amonio sustituidos.

El grupo –OH presenta un basicidad similar, aunque más débil. Ya nos hemos familiarizado con los iones oxonio, ROH2+.
![]()
Los efectos del NH2
y _OH en la sustitución electrofílica aromática pueden explicarse si suponemos
que el nitrógeno y el oxígeno comparten más de un par de electrones con el
anillo y pueden acomodar una carga positiva.
El carbocatión generado por
el ataque para con respecto al grupo NH2 de la anilina se considera,
por ejemplo, un híbrido no sólo de estructuras I, II y III, con cargas
positivas acomodadas sobre carbonos anulares, sino también de estructura IV,
donde la carga positiva la tiene el ntrógeno.
La estructura IV es particularmente
estable, porque en ella todos los átomos (excepto el hidrógeno, por supuesto)
tienen octetos electrónicos completos. Este carbocatión es mucho más estable
que el obtenido por ataque al benceno, o el que resulta por ataque meta al
grupo NH2 de la anillina (V a Vii). En ninguno de estos
casos es posible una estructura como IV. (Compárense las estabilidades de los
iones NH4+ y CH3+, por ejemplo. Aquí no se trata de cuál átomo,
nitrógeno o carbono, puede acomodar mejor
una carga positiva; es cuestión de identificar el átomo que tiene un
octeto electrónico completo.)

El examen
de las estructuras
correspondientes (VIII a XI) indica que el ataque orto es muy similar al para:
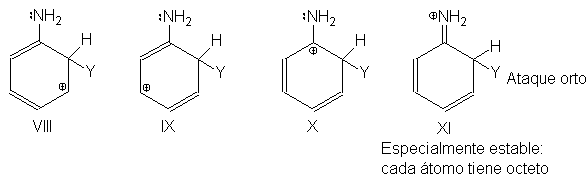
Así, la sustitución en
anilina es mucho más rápida que la sustitución en benceno, y se realiza
predominantemente en las posiciones orto y para al –NH2.
Del mismo modo se explica
la activación y orientación orto, para del grupo –OH, que se deben a la
contribución de estructuras como XII y XIII, donde todo átomo tiene un octeto
electrónico completo:
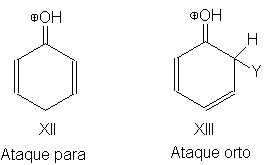
Los efectos similares de
los derivados del NH2 y OH se justifican por la contribución de
estructuras semejantes (ilustradas sólo para el ataque para):
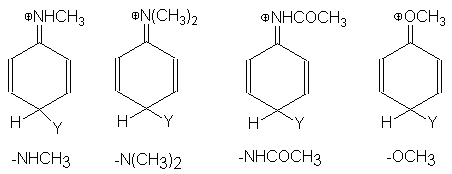
En grupos como éstos, la
tendencia del oxígeno y el nitrógeno a compartir más de un par de electrones
con un anillo aromático se comprueba en muchas a otras circunstancias
.
Gran parte de lo expuesto
recientemente nos debería parecer familiar. Habíamos visto vimos que el orbital
p lleno de un átomo de oxígeno puede solapar al orbital p vacío de un carbono
adyacente deficientemente en electrones, proporcionándole así los electrones
que necesita. Aquí se implica básicamente lo mismo, excepto que por medio del
solapamiento con el sistema n conjugado del anillo de bencenonio un elemento
como el oxígeno puede proporcionar electrones a carbonos electrónicamente
deficientes más remotos.
16.- Efecto del halógeno en la sustitución electrofílica aromática
Los halógenos son
excepcionales en cuanto a su efecto en la sustitución electrofílica aromática:
son desactivantes, sin embargo dirigen orto y para. La desactivación es
característica de la atracción electrónica, mientras la orientación orto-para
es característica de la liberación de electrones. ¿Puede el halógeno liberar y
atraer electrones?
La respuesta es afirmativa.
El halógeno atrae electrones por su efecto inductivo, y los libera por su
efecto de resonancia. Cabe suponer que los grupos NH2 y OH también pueden
actuar así, pero en este caso el efecto de resonancia, mucho más equilibrados y
se observa que operan ambos
Consideremos primero la
reactividad. El ataque electrofílico al benceno genera el carbocatión I, y el
ataque al clororbenceno, el ión
II. El efecto inductivo de atracción de
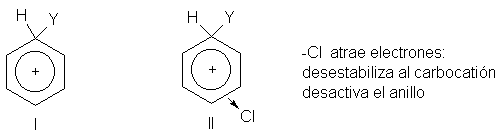
electrones que ejerce el cloro intensifica la carga positiva en el carbocatión
II, hace al ión menos estable y provoca una reacción más lenta.
Ahora, para comprender la
orientación, comparemos las estructuras de los carbocationes formados por el
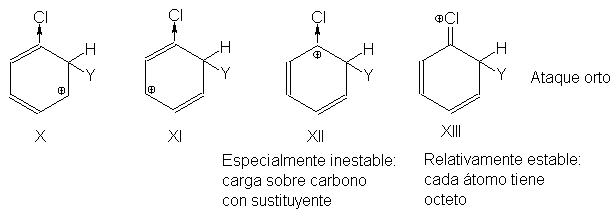
ataque en las posiciones para y meta del clorobenceno. Cada uno de ellos es un
híbrido de estas estructuras, III a V para para, VI a VIII para meta. En una de estas seis estructuras, la IV, la
carga positiva se halla ubicada en el
carbono unido al cloro. Por medio de su efecto inductivo, el cloro atrae más
intensamente los electrones del carbono unido a él, haciendo especialmente
inestable la estructura IV. Como antes, esperamos que IV contribuya poco al
híbrido, que debería ser, por tanto, menos estable que el ión híbrido que
resulta del ataque en las posiciones meta. Por
consiguiente, si sólo estuviera involucrado el efecto inductivo,
esperaríamos desactivación, además de orientación meta.

Sin embargo, la existencia
de iones halogenonio indica que un
halógeno puede compartir más de un par de electrones y acomodar una carga
positiva. Si aplicamos esta idea al problema en consideración, ¿qué
encontramos? El ión generado por el ataque en para no sólo es un híbrido de
estructuras III a V, sino también de estructura IX, donde el cloro tiene una
carga positivas y está unido al anillo por medio de un doble enlace. Esta
estructura debe ser relativamente estable, porque uno de sus átomos (salvo el
hidrógeno, por supuesto)
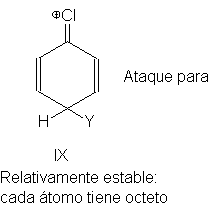
Tiene un octeto electrónico
completo. (la estructura IX es exactamente análoga a las que se propusieron para
justificar la activación y orientación orto, para originada por NH2 y –OH.)
Dicha estructura no es posible para el ión que resulta del ataque meta. La
estructura IX estabiliza el ión que resulta del ataque para en la medida en que
contribuye al híbrido, haciéndolo más estable que el generado en el ataque
meta. Aunque no podríamos haber pronosticado la importancia relativa de los dos
factores la inestabilidad de IV y la estabilización por IX, el resultado indica
que la contribución de IX es la más importante.
De la misma manera, puede
observarse que el ataque en una posición orto también da un ión (X a XIII)
estabilizado por el acomodo de la carga positiva en el cloro.
Por su efecto inductivo, el
halógeno tiende a atraer electrones, desestabilizando así el carbocatión
intermediario. Este efecto se hace
sentir para el ataque en todas las posiciones, pero es más marcado para el
ataque en las posiciones orto y para al halógeno.
Por su efecto de resonancia,
el halógeno tiende a liberar electrones, con
lo que estabiliza el carbocatión intermediario. Esta liberación sólo es
efectiva para el ataque en las posiciones orto y para el halógeno.
El efecto inductivo es más
fuerte que el de resonancia y produce
atracción neta de electrones y, por
tanto, desctivación para el ataque en todas las posiciones. El efecto de
resonancia tiende a oponerse al inductivo para el ataque en las posiciones orto
y para, por lo que aminora más la
desactivación en éstas que en meta.
De esta forma, la
reactividad está controlada por el efecto inductivo más fuerte, y la
orientación, por el de resonancia, que, aunque más débil, es más selectivo.
En la adición electrofílica
a halogenuros vinílicos observamos que el halógeno desempeña el mismo papel
dual como sustituyente en la formación de un carbocatión. También allí la
reactividad está por el efecto inductivo, y la orientación, por el de
resonancia.
Hemos establecido así que
un solo concepto estructural la formación de un doble enlace aprcial entre
halógeno y carbono contribuye a justificar las propiedades químicas poco
comunes de compuestos aparentemente tan diferentes como los halogenuros de
arilo y los de vinilo. Las estructuras que comprenden halógeno con doble
enlace, que probablemente tienen una contribución importante, no sólo par alos
iones bencenonio, sino también para los halogenuros de arilo originales (Sec.
29.6), ciretamente no parecen concordar con nuestro concepto usual de
racionalida (Sec. 10.10). Sin emabrgo, la evidencia nos obliga, por su propio
peso, a aceptar la idea de que ciertos enlaces carbono-halógeno poseen carácter
de dobles. Si esta idea nos parece extraña, simplemente demuestra que, después
de todo, sabemos muy poco sobre la estructura molecular.
14.4
Relación con otras
reacciones de carbocationes
En resumen, podemos decir
que la reactividad y la orientación en la sustitución electrofílica aromática
están determinadas por la velocidad de la formación de lso carbocationes
intermediarios. Estas velocidades son paralelas a las estabilidades de dichos
iones, que están determinadas por las
tendencias a la liberación o atracción electrónica de los grupos sustituyentes.
Un grupo puede liberar o
atraer electrones por medio de un efecto inductivo, uno de resonancia o por
ambos. Estos efectos sólo se oponen entre sí para los grupos NH2 y
OH ( y sus derivados) y para los halógenos, -X. El efecto de resonancia es el
más importante para NH2 y OH. Para X están más equilibrados: lso
halógenos son excepcionales por ser grupos desactivantes, pero son directores
orto, para.
Hemos justificado hechos en la sustitución electrofílica aromática de la misma forma que la reactividad en la sustitución SN1 y la eliminación E1; la facilidad relativa en la deshidratación de alcoholes, y la reactividad y orientación en la adición electrofílica a alquenos: cuanto más estable es el carbocatión, mayor es la velocidad de su formación; cuanto más rápido se forma el carbocatión, mayor es la velocidad de la reacción.
En todo este proceso
consideramos la estabilidad de un carbocatión sobre una misma base: la
dispersión o concentración de carga debida a la liberación o atracción de
electrones por parte de los sustituyentes. Veremos cómo el enfoque que operó
bien para la eliminación, adición y sustitución electrofílica aromática,
también funciona para la sustitución nucleofílica aromática, en la que se desarrolla una carga negativa.
Finalmente, observaremos que este tratamiento ayudará a comprender la acidez o
basicidad de compuestos como los ácidos carboxílicos, ácidos sulfónicos, aminas
y fenoles.