Alquinos
(Ir a página principal http://organica1.org/ )
2 Estructura del acetileno. El triple enlace
carbono-carbono
3 Alquinos superiores. Nomenclatura
4 Propiedades físicas de los alquinos
5 Fuente industrial del acetileno
9 Adición electrofílica a alquinos
10 Hidratación de alquinos. Tautomería.
11 Acidez de los alquinos. Acidos muy débiles
12 Reacciones de los acetiluros metálicos.
Síntesis de alquinos
13 Formación de enlaces carbono-carbono.
1
Introducción
Hasta ahora hemos tratado
dos tipos de enlace carbono-carbono: el enlace simple y el doble.
El enlace simple
carbono-carbono es de reactividad baja; su función principal es la de actuar
como cemento para mantener unidos casi todos los compuestos orgánicos. El doble
enlace carbono-carbono es insaturado y, en consecuencia, muy reactivo frente a
una amplia variedad de reactivos; como sustituyente, puede ejercer efectos
notables sobre el resto de la molécula.
Llegamos de esta manera, al
triple enlace carbono-carbono, el grupo funcional de la familia denominada
alquinos. Al igual que el doble enlace, es insaturado y altamente reactivo:
frente a los reactivos con los que reaccionan los enlaces dobles y hacia otros.
También puede ejercer una influencia notable en el resto de la molécula, y de
una forma peculiar. Por medio de una combinación de sus propiedades
características, el triple enlace carbono-carbono tiene una función especial de
importancia cada vez mayor en la síntesis orgánica.
2
Estructura del acetileno. El triple enlace carbono-carbono
El miembro más sencillo de
la familia de los alquinos es el acetileno, C2H2. Empleando
los métodos que aplicamos a la estructura del etileno, obtenemos una estructura
en la que los átomos de carbono comparten tres pares de electrones; es decir,
están unidos por un triple enlace. El triple enlace carbono-carbono es la
característica distintiva de la estructura de los alquinos.
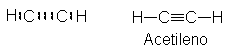
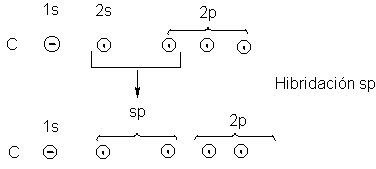
Una vez más, la mecánica
cuántica da mucha más información sobre
el acetileno y su triple enlace. Para formar enlaces con dos átomos
adicionales, el carbono utiliza dos orbitales híbridos equivalentes: orbitales
sp que resultan de mezclar un orbital s
y otro p. Estos orbitales sp se encuentran en una línea recta que pasa por el
núcleo del carbono, por lo que el ángulo entre ellos es de 180º. Esta disposición
lineal permite la máxima separación de
los orbitales híbridos. Así como la repulsión entre orbitales genera cuatro
enlaces tetraédricos o tres trigonales, también genera dos enlaces lineales.
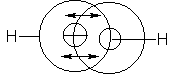
Si ordenamos los dos
carbonos y los dos hidrógenos de manera que se produzca un solapamiento de
orbitales, obtenemos la estructura mostrada en la figura 1.
![]()
El acetileno es una
molécula lineal que tiene los cuatro átomos ubicados a lo largo de una línea
recta. Tanto los enlaces carbono-hidrógeno como los carbono-carbono son
cilíndricamente simétricos en torno a una línea que une los núcleos; por tanto,
son enalces o.
Sin embargo, la molécula
aún no está completa. Al formar los orbitales sp recién descritos, cada carbono
sólo utiliza uno de sus tres orbitales p, por lo que aún quedan otros dos. Cada
uno de estos últimos consta de dos lóbulos iguales, cuyo eje es perpendicular,
al eje del otro orbital p y al de los orbitales sp; cada orbital p está ocupado
por un solo electrón. La suma de dos orbitales p perpendiculares no da cuatro
lóbulos esféricos, sino una sola nube con forma de rosca (Fig. 2). El
solapamiento de los orbitales p de un carbono
Con los del otro permite el
apareamiento de electrones. Se forman dos enlaces p, que juntos generan una envoltura cilíndrica
en torno a la línea de unión de los núcleos (Fig. 3). Por tanto, el triple
enlace carbono-carbono está constituido por un enlaces o fuerte y dos p más
débiles; su energía total es de 198 kcal. Es más fuerte que el doble enlace del
Etileno (163 kcal) o
el simple del etano (88 kcal), por
consiguiente es más corto que ambos.
Nuevamente, esta estructura
de la mecánica cuántica se comprueba directamente. La difracción electrónica, la
de rayos X y la espectroscopia indican que el acetileno (Fig. 4) es
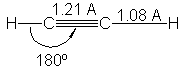
Una molécula lineal. La
distancia C-C es 1.21 A, comparada con 1.34 A del etileno y 1.53 A del etano.
En la figura 5 se muestra un modelo de la molécula de acetileno.
Como en el caso del doble
enlace, la estructura del triple enlace queda verificada aunque esta vez de
forma negativa por la evidencia del número de isómeros. Los modelos nos
permiten apreciar que la linealidad de los enalces no admite isomería
geométrica: tales isómeros no han sido encontrados.
En el acetileno, la
distancia C-H es 1.08 A, aún más corta que en el etileno (1.103 A); debido a su
mayor carácter s, los orbitales sp son más pequeños que los sp2, por
lo que el carbono con hibridación sp forma enlaces más cortos que el carbono
con hibridación sp2.
No se conoce la energía de
disociación del enlace C-H del acetileno, pero cabe suponer que es mayor que la
del etileno. Extrañamente, la misma hibridación sp, que con toda seguridad hace
más dificil la ruptura del enlace C-H para formar radicales libres (homólisis),
facilita, como veremos (Sec. 11), la ruptura para formar iones (heterólisis).

3
Alquinos superiores. Nomenclatura
Al igual que los alcanos y
alquenos, los alquinos forman una serie
homóloga, también con un incremento de CH2.
Los alquinos se denominan
de acuerdo con dos sistemas. Según uno, se les considera como derivados del
acetileno, por reemplazo de uno o ambos átomos de hidrógeno por grupos alquilo.
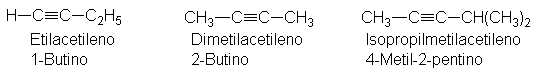
Para alquinos más
complicados se utilizan los nombres
IUPAC. Las reglas son las mismas que las empeladas para denominar los
alquenos, salvo que la terminación eno se reemplaza por ino. La estructura de
referencia es la cadena continua más larga que contenga el triple enlace, y las
posiciones de los sustituyentes y del triple enlace se indican con números. A
este último se le da el número del primer carbono triplemente enlazado,
contando desde el extremo de la cadena más cercano.
4
Propiedades físicas de los alquinos
Al ser compuestos de baja
polaridad, las propiedades físicas de los alquinos son, en esencia, las mismas
que las de los alcanos y alquenos. Son insolubles en agua, pero bastante
solubles en disolventes orgánicos usuales
y de baja polaridad: ligroína, éter, benceno, tetracloruro de carbono. Son
menos densos que el agua y sus puntos de ebullición (Tabla 11.1) muestran el
aumento usual con el incremento del número de carbonos y el efecto habitual de
ramificación de las cadenas. Los puntos de ebullición son casi los mismos que
para los alcanos o alquenos con el mismo esqueleto carbonado.
5
Fuente industrial del acetileno
El alquino industrial más
importante es el miembro más simple de la familia, el acetileno.
Tradicionalmente, se ha preparado por la acción del agua sobre carburo de
calcio, CaC2, que se obtiene, a su vez, por reacción entre el óxido
de calcio y el coque, a las altísimas temperaturas del horno eléctrico. Por su
parte, el óxido de calcio y el coque se consiguen de la caliza y del carbón,
respectivamente.
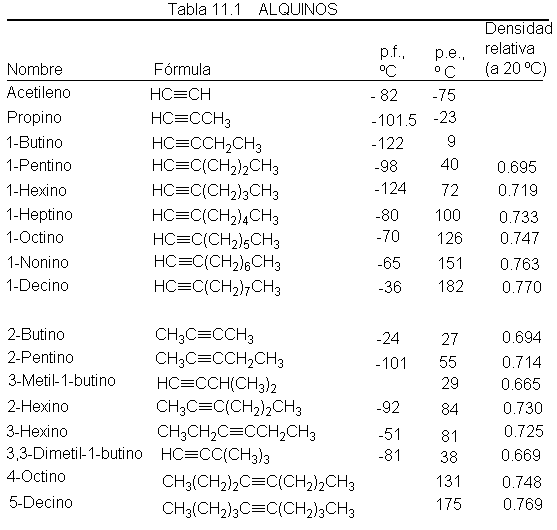
Una síntesis alternativa,
basada en el petróleo, está desplazado al proceso del carburo.
Implica la oxidación
parcial, controlada y a temperatura elevada, del metano.
![]()
Debido al elevado costo del
acetileno, ha disminuido mucho su antiguo y vasto mercado; la mayoría de los
productos químicos que antes sintetizaban del acetileno se obtienen en la
actualidad del etileno. No obstante, el acetileno sigue siendo la fuente de
algunos compuestos utilizados en la manufactura de polímeros.
6
Preparación de alquinos
La síntesis de alquinos
puede implicar dos procesos: la generación de un triple enlace carbono-carbono,
o el aumento del tamaño de una molécula que ya contiene un triple enlace.

Un triple enlace
carbono-carbono se genera del mecanismo modo que uno doble: por eliminación de
átomos o grupos en dos carbonos adyacentes. Los grupos que se eliminan, y los
reactivos que se emplean son esencialmente los mismos que en la preparación de
los alquenos.
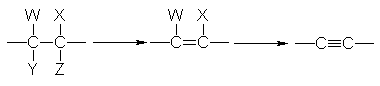
La deshidrogenación de
dihalogenuros vecinales es particularmente útil, puesto que los propios
dihalogenuros se obtienenn con facilidad de los alquenos correspondientes por
adición de halógeno. Esto significa la conversión en varias etapas de un doble
enlace en uno triple.
Por lo general, la
deshidrohalogenación puede realizarse en dos etapas, tal como se indica:
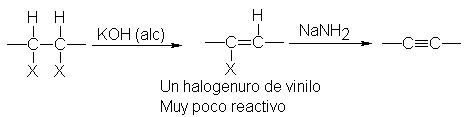
La primera etapa de la
reacción es un método valioso para preparar halogenuros no saturados. Los
halogenuros obtenidos así, con halógeno unido directamente al carbono con el
doble enlace, se denominan halogenuros vinílicos, y son muy inertes. Por tanto,
en condiciones moderadas, la deshidrogenación se detiene en la etapa del
halogenuro de vinilo; para la formación del alquino se precisan condiciones más
enérgicas: usar una base más fuerte.
La conversión de alquinos
pequeños en otros más grandes se logra utilizando acetiluros metálicos, que son
particularmente fáciles de generar debido a una propiedad especial de ciertos
alquinos. Una vez sintetizados, resultan ser reactivos muy versátiles.
7
Reacciones de alquinos
Al igual que la química de
los alquenos es la del doble enlace carbono-carbono, la de los alquinos es la
del triple enlace carbono-carbono. Como los alquenos, los alquinos sufren
adición electrofílica, y por la misma razón: la disponibilidad de los
electrones n sueltos. La adición de hidrógeno, halógenos y halogenuros de
hidrógeno a los alquinos es muy similar a la adición a los alquenos, salvo que
en este caso pueden consumirse dos moléculas de reactivo por cada triple enlace. Como se ha mostrado, si se
eligen condiciones apropiadas, es posible limitar la reacción a la primera
etapa aditiva: la formación de alquenos. En algunos casos al menos, esto se simplifica
debido a que la forma de introducir los sustituyentes en la primera etapa
afecta a la segunda.
Además de la adición, los
alquinos dan ciertas reacciones que se deben a la acidez de un átomo de
hidrógeno unido a un carbono con triple enlace.

8 reducción a alquenos
La reducción de un alquino
hasta la etapa del doble enlace puede dar un cis- o un trans- alqueno, a menos que
el triple enlace se encuentra en un extremo de la cadena. El isómero
predominante que resulte depende de la elección del agente reductor.
Por reducción de alquinos
con sodio o litio en amoniaco líquido se obtiene, predominantemente, el
trans-alqueno. Puede lograrse cis-alqueno casi
puro (hasta un 98%) por hidrogenación de alquinos con varios
catalziadores diferentes, entre ellos con un paladio preparado especialmente
conocido como catalizador de Lindlar.
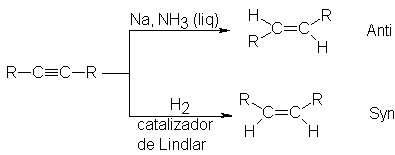
En consecuencia, cada una
de estas reacciones es fuertemente estereoselectiva. De manera general, se
atribuye esta estereoselectividad en la reducción syn de alquinos a la unión de
dos átomos de hidrógeno al mismo lado de un alquino situado en la superficie
del catalizador; es de suponer que esta misma estereoquímica vale para la
hidrogenación de alquinos terminales, RC=CH, que no pueden dar alquenos cis y
trans.
Aquí no se tratará el
mecanismo que da lugar a la reducción anti.
Varias veces hemos tratado
la estereoespecificidad: organismos que discriminan entre isómeros geométricos.
Vimos algunos ejemplos de esto en la respuesta de insectos hacia a las
feromonas . Para que los materiales sintéticos sean efectivos en organismos
vivos, la estereoespecificidad de la acción biológica exige una estereoselectividad
en la síntesis de estos materiales. Se han creado nuevos métodos para generar
selectivamente dobles enlaces, pero el más sencillo, y a la vez el más
socorrido, es la hidrogenación de alquinos.
El tema va más allá, sin
embargo. Estos alquenos pueden ser los productos finales deseados, como sucede
con algunas feromonas, pero a menudo son una etapa intermedia. Los alquenos
sufren varias reacciones, muchas de ellas diastereoselectivas e incluso
enantioselectivas; si ha de utilizarse esta estereoselectividad a fondo, debe
comenzarse con un alqueno estereoquímicamente puro.
9
Adición electrofílica a alquinos
La adición de ácidos como
los halogenuros de hidrógeno es electrofílica, y aparentemente sigue el mismo
mecanismo con los alquinos que con los alquenos; es decir, por la vía de un
carbocatión intermediario. La diferencia estriba en que ahora el intermediario
es un catión vinílico.
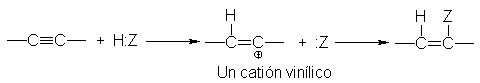
Habíamos visto que los cationes vinílicos son aún menos estables
que los alquílicos primarios, en relación con los sustratos para heterólisis;
vimos, también, que su formación por heterólisis; vimos, también, que su
formación por heterólisis es comparativamente lenta, pudiendo generarse sólo
por la salida de grupos salientes super.
Ahora bien, comentamos que en la adición electrofílica a alquenos,
la reactividad depende de la
estabilidad del carbocatión intermediario: cuanto más estable es éste, mayor es
la velocidad de su formación. ¿Significa esto, entonces, que la adición a los alquinos es considerablemente más
lenta que a los alquenos?
No es mucho más lenta: la
adición de ácidos próticos a los alquinos sucede a casi la misma velocidad que a los alquenos. La
explicación se encuentra en nuestra definición de la estabilidad de un
carbocatión: La explicación se encuentra en nuestra definición de la
estabilidad de un carbocatión: la relación con el sustrato del cual es
generado. En comparación con los sustratos para heterólisis, los cationes
vinílicos son inestables y hemos atribuido esto al enlace extraordinariamente
fuerte que sujeta al grupo saliente en los sustratos vinílicos: no a ninguna inestabilidad inherente en los
propios cationes. Los cationes vinílicos son lentos en formarse por
heterólisis; sin embargo, en reacciones de adición, los sustratos son alquenos
y alquinos; estos compuestos deben
servir de estándares de comparación para las estabilidades de carbocationes: un
alqueno para un carbocatión saturado y un alquino para uno vinílico. En lo referente
al sustrato del cual se genera cada uno en una reacción de adición, ambos son de estabilidad similar. La subida
energética de un alquino a un catión vinílico es aproximadamente la misma que
para un alqueno hasta un catión saturado.
Con respecto a la adición
de halógenos, los alquinos son considerablemente menos reactivos que los
alquenos. Para los alquenos, vimos que esta reacción implica la formación
inicial de un ion halogenonio cíclico. Se ha atribuido la reactividad menor de
los alquinos a la mayor dificultad de formación de tales intermediarios
cílcicos.
10
Hidratación de alquinos. Tautomería.
Al igual que los alquenos,
los alquinos pueden hidratarse. En presencia de un ácido – y para alquinos
simples, también HgSO4 se añade una molécula de agua al triple
enlace. Esto implica adición electrofílica, como en la hidratación de alquenos,
y procede mediante carbocationes. Sin embargo, a primera vista esto no parece
ser así.
Consideremos el ejemplo más
sencillo: la hidratación del propio acetileno. El producto obtenido es
acetaldehído, CH3CHO, que parece ser extraño, si se consideran los
grupos que se ligan a los dos carbonos del triple enlace. Sin embargo, el
producto tiene una explicación bastante sencilla.
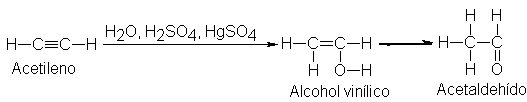
Si la hidratación del
acetileno siguiera el mismo esquema que la de los alquenos, se obtendría una
estructura que llamaríamos alcohol vinílico, por adición de H y OH al triple
enlace. Sin embargo, todos los intentos por separar este alcohol tienen como
resultado al igual que la hidratación del acetileno la formación de
acetaldehído.
Una estructura con OH unido
a un doble enlace carbono-carbono se denomina enol (eno, por dicho doble enlace
carbono-carbono, y ol, por alcohol). Por lo general es cierto que cuando
intentamos preparar compuestos con la estructura de un enol, obtenemos en
cambio una estructura cetónica (una que contiene un grupo C=O). Hay un
equilibrio entre ambas estructuras, pero suele favorecer mucho más a la forma
cetónica. Así, se forma inicialmente alcohol vinílico por hidratación del
acetileno, pero se convierte rápidamente en una mezcla equilibrada que consiste
casi por completo en acetaldehído.
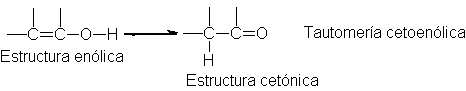
Las transposiciones del
tipo ceto-enol se producen con mucha facilidad, debido a la polaridad del
enlace O-H. Un ion hidrógeno se prepara sin dificultad del oxígeno para generar
un anión híbrido, pero cuando vuelve un ion hidrógeno (seguramente uno
diferente), puede unirse al oxígeno o al carbono del anión. Si vuelve al
oxígeno, puede separarse de nuevo sin dificultad, pero si se combina con el
carbono, tiende a quedarse allí, lo que representa un ejemplo de la tendencia
de un ácido fuerte a desplazar de sus sales a uno más débil .
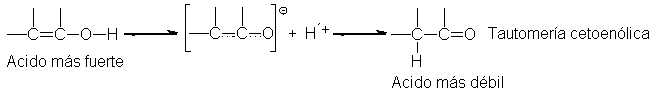
Los compuestos cuyas estructuras
difieren marcadamente en la disposición de sus átomos, pero que existen en un
equilibrio rápido y fácil, se denominan tautómeros. El tipo más común de
tautomería comprende estructuras que difieren en el punto de unión del
hidrógeno; en estos casos, como en la tautomería cetoenólica, el equilibrio
tautómero favorece generalmente a la estructura con el hidrógeno unido al
carbono, en vez de a un elemento más electronegativo; o sea, favorece al ácido
más débil.
11
Acidez de los alquinos. Acidos muy débiles
En nuestro estudio inicial
de los ácidos (en el sentido empleado por Lowry-Bronsted ),consideramos la
acidez como una medida de la tendencia de un compuesto a perder un ion
hidrógeno. Los compuestos que por lo común muestran acidez apreciable son los
que tienen hidrógeno unido a un elemento bastante electronegativo (N, O, S, X,
por ejemplo). El enlace que sujeta al
hidrógeno es polar, por lo que este hidrógeno, relativamente positivo, puede
separarse en forma de catión; desde otro punto de vista, un elemento
electronegativo puede acomodar mejor el par de electrones abandonado. De
acuerdo con la serie de electronegatividades, F >O >N> C, no es
extraño que el HF sea un ácido bastante fuerte, el H2O es relativamente débil,
el NH3 es aún más débil y el CH4 es tan débil que
normalmente no se considera como un ácido.
En la química orgánica nos
ocupamos frecuentemente de la acidez de sustancias que no enrojecen al tornasol, ni neutralizan bases acuosas, sin
embargo tienen tendencia aunque pequeña a perder un ion hidrógeno.
Un carbono con triple
enlace se comporta como si fuera un elemento enteramente diferente más
electronegativo que el carbono con enlace simple o doble. Como consecuencia, un
hidrógeno unido a un carbono con triple enlace, como en el acetileno o en
cualqueir alquino con triple enlace en el extremo de la cadena (RC=C-H),
presenta una acidez apreciable. Por ejemplo, el sodio reacciona con acetileno
para liberar gas hidrógeno y formar el compuesto acetiluro de sodio.
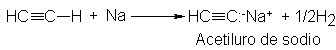
¿Cuál es la aciedez del
acetileno? Comparémoslo con dos compuestos familiares, amoniaco y agua.
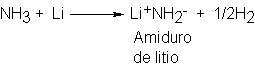 El litio metálico reacciona con amoniaco para
formar amiduro de litio, LiNH2, que es la sal del ácido débil H-NH2.
El litio metálico reacciona con amoniaco para
formar amiduro de litio, LiNH2, que es la sal del ácido débil H-NH2.
La adición de acetilenoa amiduro
de litio disuelto en éter, forma amoniaco y acetiluro de litio.
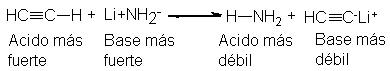
El ácido más débil, H-NH2,
es desplazado de su sal por el ácido más fuerte HC=C-H.
En otras palabras, la base fuerte, NH2-, arranca el protón de la base más débil, HC=C-; si el NH2- sujeta al protón más fuertemente que el HC=C-, entonces el H-NH2 debe ser un ácido más débil que el HC=C-H.
Si se agrega agua al acetiluro de litio se forma hidróxido de litio y se regenera el acetileno.
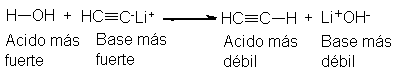
El ácido más débil, HC=C-H,
es desplazado de su sal por el ácido más fuerte, H-OH.
Se ve así que el acetileno
es un ácido más fuerte que el amoniaco, pero más débil que el agua.
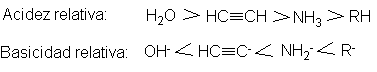
Otros alquinos con
hidrógeno unido a un carbono de triple enlace es decir, los alquinos terminales
muestran una acidez comparable.
El método que acabamos de
describir para comparar la acidez de los alcanos, el amoniaco y el agua es
general y se utiliza para determinar la acidez relativa de varios ácidos muy
débiles. Se demuestra que un compuesto es un ácido más fuerte que otro por su
capacidad para desplazar de sus sales al segundo.
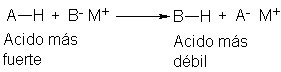
Según nuestra secuencia, el
acetileno debe ser un ácido más fuerte que un alcano, RH.
Eso es cierto, y la diferencia en acidez es de
utilidad considerable en síntesis. Si se trata un acetileno terminal con un
halogenuro de alquilmagnesio o un alquil-litio, se desplaza al alcano de su sal
y se obtiene el acetiluro metálico. Por ejemplo,
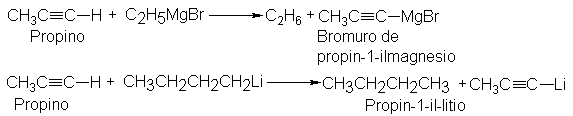
Estas reacciones muestran
el mejor camino hacia esos importantes compuestos organometálicos.
¿Cómo podemos justificar el
hecho de que el hidrógeno ligado a un carbono de triple enlace sea
especialmente ácido? ¿Cómo podemos explicar que el acetileno sea un ácido más
fuerte que el etano, por ejemplo? Una interpretación posible puede hallarse en
las configuraciones electrónicas de los aniones.
Si el acetileno es un ácido
más fuerte que el etano, entonces el ion acetiluro debe ser una base más débil
que el ion etiluro, C2H5-. En el anión acetiluro, el par
no compartido de electrones ocupa un orbital sp; en el anión etiuro, el par de
electrones no compartido ocupa un orbital sp3. La disponibilidad de este par
para ser compartido con ácidos determina la basicidad del anión. En comparación
con un orbital sp3, uno sp tiene menos carácter p y más s . Un electrón en un
orbital p se encuentra a cierta distancia del núcleo y está relativamente
suelto; en cambio, un electrón en un orbital s se halla cerca del núcleo, y
está sujeto más firmemente. El ion acetiluro es la base más débil, pues su par
de electrones, ubicado en un orbital sp, está sujeto más firmemente.
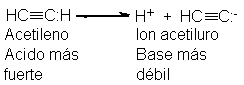
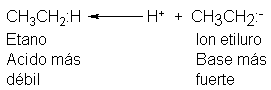
12
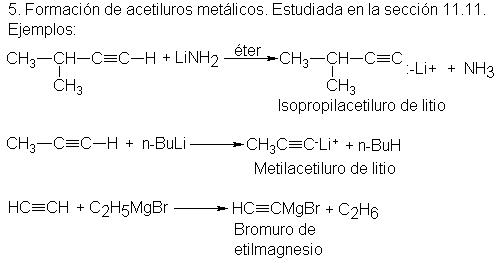
Reacciones de los acetiluros metálicos. Síntesis de alquinos
¿Por qué describimos los
acetiluros metálicos como compuestos organometálicos importantes? Porque nos
permite convertir alquinos pequeños en otros más grandes.
Al igual que los
cuprodialquil-litios los acetiluros de litio y sodio pueden reaccionar con halogenuros de alquilo primarios.
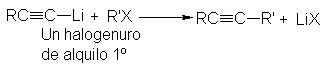
El grupo alquilo se une al
carbono del triple enlace y se genera un alquilo nuevo más largo.
Por ejemplo,
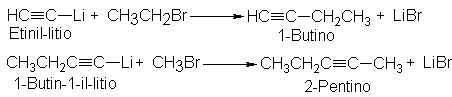
Esta reacción sólo da
rendimientos aceptables con halogenuros de alquilo primarios, y por una razón ya
mencionada: los iones acetiluro son bases fuertes, de modo que con halogenuros
de alquilo secundarios o terciarios la reacción predominante es la eliminación.
Como veremos en los capítulos 17 y 18, los alcoholes complicados pueden
sintetizarse fácilmente utilizando reactivos de Grignard y compuestos
organolitiados. Dichas síntesis puede involucrar tanto organometálicos
alquílicos como sus contrapartes alquinílicas, generando así moléculas que
contienen dos grupos muy reactivos, el
triple enlace carbono-carbono y el OH.
13
Formación de enlaces carbono-carbono.
Función de los compuestos organometálicos
El tipo de síntesis
descrito anteriormente tiene algo muy especial. Tomamos dos moléculas orgánicas
y las convertimos en otra más grande. Hemos realizado algo que se halla en el
centro de la síntesis orgánica: hemos establecido un enlace carbono-carbono.
Veamos más de cerca este proceso y la función especial de los compuestos
organometálicos.
Los enlaces carbono-carbono
se generan más comúnmente en forma heterolítica, lo que significa que uno de
los carbonos proporciona un par de electrones, y el otro lo acepta; es decir,
hay una reacción entre un carbono nucleofílico y otro electrofílico.
Exceptuando el hidrógeno u
otro carbono, los elementos que encontramos generalmente ligados a un carbono
son más electronegativos que éste, por lo que le arrancan electrones: halógeno
en los halogenuros de alquilo, por ejemplo, o, como veremos, oxígeno en
aldehídos y cetonas. En tales compuestos, el carbono es electrónicamente
deficiente y, en consecuencia, electrofílico, por lo que tiende a reaccionar
con nucleófilos. Por esto, la sustitución nucleofílica es típica de los
halogenuros de alquilo; para los aldehídos y cetonas es típica la adición
nucleofílica.
Para que estas reacciones
den lugar a la formación de un enlace carbono-carbono, debemos utilizar
reactivos donde el elemento nucleófilo sea carbono. ¿Dónde se pueden encontrar
esos reactivos? En los compuestos organometálicos. Al igual que los elementos
electronegativos hacen electrofílico al carbono, los elementos electropositivos
los metales lo comvierten en nucleofílico. La reacción entre el carbono
nucleófilo de un reactivo organometálico y el carbono electrófilo de un
sustrato es la que da origen a un nuevo enlace carbono-carbono.
Los compuestos
organometálicos se sintetizan comúnmente a partir de halogenuros orgánicos; en
esta síntesis se modifica la naturaleza del carbono, de electro- a
nucleofílica. Esta reacción es quizás el ejemplo más antiguo y simple de lo que
se llama umpolung; esto es, la inversión de la polaridad del carbono. El
concepto de umpolung se aplica actualmente de diversas maneras, en un esfuerzo
por crear un carbono nucleofílico. En la formación de un compuesto
organometálico, los electrones que enriquecen al carbono proceden en definitiva
del metal libre, que hace por naturaleza lo normal en los metales; liberar
electrones.
En la formación de enlaces
carbono-carbono hemos utilizado reactivos organometálicos: la síntesis de
Corey-House para hidrocarburos. En comparación con esta reacción, el uso de
acetiluros metálicos tiene una ventaja especial: no sólo se genera un nuevo
enlace carbono-carbono, sino que el producto contiene un grupo funcional muy
reactivo.
No es de extrañar que el
triple enlace carbono-carbono se haya convertido en parte importante de la
síntesis orgánica. La acidez de un alquino terminal permite su fácil conversión en un acetiluro metálico.
Mediante este acetiluro, es psoible introducir una unidad estructural con
triple enalce en moléculas de numerosos tipos. Este triple enlace puede
convertirse por adición en mcuhos otros compuestos. En particular en un doble
enlace y con alto grado de estereoselectividad. Tenemos ahora un doble enlace
de estereoquímica conocida en una ubicación específica de la molécula, quedando
abierta la puerta a las múltiples reacciones que tienen lugar en este grupo
funcional.
14 Análisis de los
alquinos
Los alquinos responden a las
pruebas de caracterización igual que los alquenos: decoloran al bromo en
tetracloruro de carbono sin desprendimiento de bromuro de hidrógeno y decoloran
el permanganato diluido,, neutro y frío; no son oxidados por el anhídrido
crómico. Como los dienos, sin emabrgo, son más insaturados que los alquenos,
propiedad que puede
Detectarse por
determinación de sus fórmulas moleculares (CnH2n-2) y por
una hidrogenación cuantitativa (se consumen dos moles de hidrógeno por mol de
hidrocarburo).
La comprobación de la
estructura se hace mejor por los mismos métodos de degradación empleados en el
estudio de los alquenos. Por ozonólisis, los alquinos dan ácidos carboxílicos,
mientras que los alquenos dan aldehídos y cetonas. Por ejemplo:
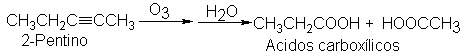
Los alquinos ácidos reaccionan
con ciertos iones de metales pesados, principalmente con Ag+ y Cu+, para formar
acetiluros insolubles. La formación de un precipitado al añadir un alquino a
una solución de AgNO3 en alcohol, por ejemplo, es un indicio de hidrógeno unido a un carbono con triple
enlace. Esta reacción puede utilizarse para diferencias alquinos terminales
(con el triple enlace en el extremo de la cadena) de los no terminales.
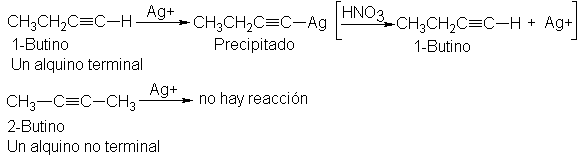
Si se permite que estos
acetiluros de metales pesados se sequen, es posible que exploten: deben ser
destruidos mientras aún estén húmedos, calentándolos con ácido nítrico; el
ácido mineral fuerte regenera al ácido débil, al acetileno.