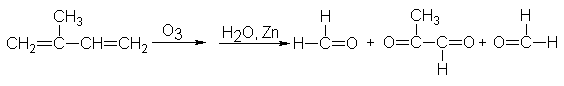Conjugación y resonancia
(Ir a página principal http://organica1.org/ )
Dienos
1 El doble enlace carbono-carbono como
sustituyente
2 Halogenación de los alquenos por radicales
libres:
3 Sustitución en alquenos por radicales
libres:
4 Sustitución en alquenos por radicales
libres:
7 El radical alilo como híbrido de
resonancia.
8 Estabilidad del radical alilo
9 Descripción orbital del radical alilo
10 Aplicación de la teoría de la resonancia
11 Estabilización por resonancia de los radicales alquilo.
12 El catión alilo como híbrido de
resonancia.
13 Sustitución nucleofílica en sustratos
alílicos: SN1.
14 Estabilización de carbocationes:
15 Estabilización de carbocationes:
16 Estabilización por resonancia de cationes
alquilo:
17 Sustitución nucleofílica en sustratos
alílicos. SN2
18 Sustitución nucleofílica en sustratos
vinílicos
19 Sustitución nucleofílica en sustratos
vinílicos: cationes vinílicos
20 Dienos: estructura y propiedades
21 Estabilidad de dienos conjugados.
22 Resonancia en dienos conjugados
23 Resonancia en alquenos. Hiperconjugación
24 Estabilidad de dienos y alquenos:
25 Facilidad de formación de dienos
conjugados:
26 Adición electrofílica a dienos conjugados.
Adición 1,4
27 Adición 1,2 contra 1,4. Velocidad contra
equilibrio
28 Adición de radicales libres a dienos
conjugados: orientación
29 Adición de radicales libres a dienos
conjugados: reactividad
30 Polimerización de dienos por radicales
libres.
31 Isopreno y la regla isoprénica
1
El doble enlace carbono-carbono como sustituyente
En el capítulo 8 comenzamos
nuestro estudio de la química del doble enlace carbono-carbono. Se imaginó este
doble enlace como un lugar en la molécula del alqueno donde puede suceder una
reacción: una adición electrofílica o
de radicales libres. Pero ésta solo es una parte de la historia. Además de dar
cabida a la adición, el doble enlace ejerce efectos poderosos sobre ciertas
reacciones que se desarrollan en otra parte de la molécula. Aunque no sufre un
cambio permanente, este doble enlace es importante en la determinación del
curso de la reacción. Esta parte de la química de los alquenos es la que
estudiaremos en este capítulo: el doble enlace carbono-carbono no como grupo
funcional, sino como sustituyente.
Hasta aquí hemos analizado
diversas familias de compuestos: alcanos, halogenuros de alquilo (y sustancias
relacionadas), alcoholes y alquenos. Hemos visto algunas de las propiedades
químicas asociadas con el grupo funcional de cada una de estas familias: C-H de
los alcanos, X y OH de los halogenuros de alquilo y alcoholes, el doble enlace
de los alquenos. Nuestro enfoque nos ha conducido a varios tipos, los más
importantes, de reacciones orgánicas: sustitución con radiales libres,
sustitución nucleofílica, eliminación y adición. Hemos tratado los efectos
de los sustituyentes sobre estas reacciones principalmente grupos alquilo: sus
efectos polares y estéricos, y los efectos (hasta el momento no especificados)
sobre la estabilidad de radicales libres y alquenos-. También vimos el efecto
inductivo de los halógenos.
En este capítulo volveremos a estudiar estas familias de compuestos y cada uno de estos tipos de reacciones, y observaremos los efectos ejercidos por un tipo diferente de sustituyente: el doble enlace carbono-carbono. Descubriremos que éste ejerce su efecto de manera distinta a la de un grupo alquilo y, como resultado, estos efectos suelen ser más poderosos. La mayor parte de estos efectos se derivan de una característica estructural denominada conjugación: la ubicación de un orbital n de manera tal que se puede solapar con otros orbitales de la molécula. Además, para reforzar nuestro planteamiento de la conjugación, emplearemos la teoría estructural conocida como resonancia.
2
Halogenación de los alquenos por radicales libres:
sustitución y
adición.
Observemos la estructura
del alqueno simple, propileno. Contiene un doble enlace carbono-carbono, en el
que tiene lugar las mismas reacciones que son características del etileno. Con
cloruro de hidrógeno, por ejemplo, el propileno sufre adición electrofílica:
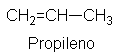
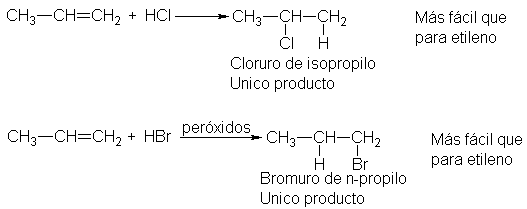
Con bromuro de hidrógeno,
en presencia de peróxidos, la adición es por radicales libres.
Pero el propileno también presenta
un grupo metilo, que modifica las reacciones que se llevan a cabo en el doble
enlace. Debido a este grupo metilo, la adición electrofílica es más rápida que
con etileno, y da exclusivamente cloruro de isopropilo. También, debido al
grupo metilo, la adición de radicales libres es más rápida que con etileno,
dando exclusivamente bromuro de n-propilo. Así, el grupo metilo afecta, como
sustituyente, a la reactividad del doble enlace carbono-carbono y determina la
orientación del ataque.
Modifiquemos ahora nuestro
enfoque y consideremos el grupo metilo, no como sustituyente, sino como lugar
de la reacción. ¿Qué tipo de reacción podemos esperar que suceda aquí? Este
grupo tiene una estructura similar a un
alcano, por lo que podemos suponer que debe dar reacciones del tipo de las de
un alcano: por lo que podemos suponer que debe dar reacciones del tipo de las
de un alcano: la sustitución de un halógeno por radicales libres, por ejemplo,
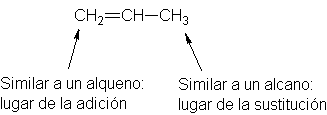
Consideremos, entonces, la
reacción del propileno con los halógenos. Sin embargo, la molécula del
propileno presenta dos sitios donde puede atacar el halógeno, el doble enlace y
el grupo metilo. ¿Podemos dirigir el ataque a sólo uno de estos lugares? La
respuesta es afirmativa: mediante nuestra elección de las condiciones de
reacción.
Sabemos que los alcanos
sufren sustitución por halógeno a temperaturas elevadas o por la influencia de
la luz ultravioleta, y generalmente en fase gaseosa: condiciones que favorecen
la formación de radicales libres. Sabemos que los alquenos se someten a
reacciones de adición a bajas temperaturas y en ausencia de luz, y generalmente
en fase líquida: condiciones que favorecen reacciones heterolíticas o que al
menos no ayudan a la generación de radicales.
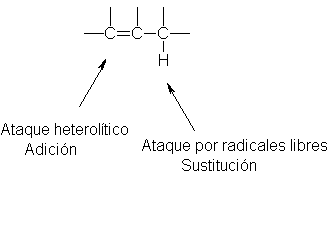
Si se desea dirigir el
ataque del halógeno a la parte
alquílica del alqueno, debemos elegir condiciones favorables para la
reacción de radicales libres, y desfavorables para la heterolítica. Los
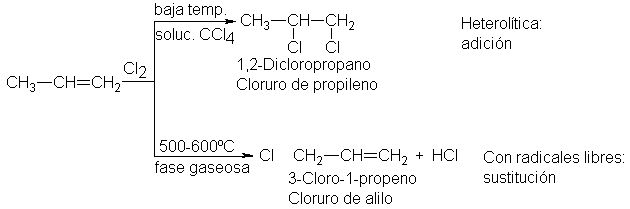
químicos de la Shell Development Company descubrieron que a temperaturas de
500-600ºC, una mezcla de propileno y cloro gaseoso da principalmente el
producto de sustitución, 3-cloro-1-propeno, conocido como cloruro de alilo (CH2 = CH – CH2- = alilo).
El bromo se comporta de
forma análoga.
Según lo expuesto antes
cabe preguntarse por qué el halógeno no se agrega al doble enlace, en lugar de
extraer un átomo de hidrógeno. H. C. Brown (Universidad de Purdue) ha sugerido
que el átomo de halógeno se adiciona, pero a temperaturas elevadas es expulsado
antes de que pueda ocurrir la segunda etapa de la adición de radicales libres.
Concuerda con la exhibición
de Brown el descubrimiento de que puede utilizarse una concentración baja de
halógeno, en vez de una temperatura elevada para favorecer la sustitución a expensas
de la adición (de radicales libres). La adición del átomo
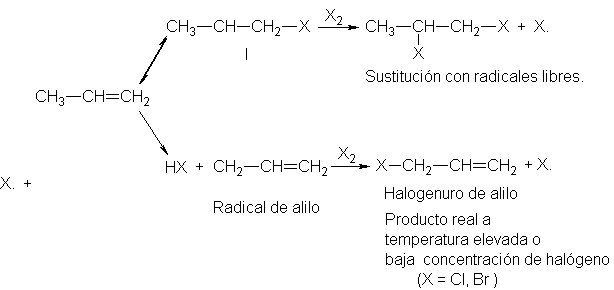
de halógeno da el radical I, que se descompone (para generar el
material de partida) si la temperatura es elevada o si no encuentra pronto una molécula
de halógeno para completar la adición. Por otra parte, una vez formado el
radical alilo, éste no tiene otra opción que esperar una molécula de halógeno,
cualquiera que sea la temperatura o lo baja que sea la concentración de
halógeno.
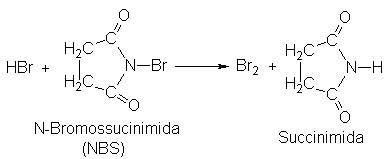
El compuesto
N-bromosuccinimida (NBS) es un reactivo empleado con el propósito específico de
bromar alquenos en la posición alílica: el NBS funciona simplemente
proporcionando una baja concentración constante de bromo. A medida que va
formándose cada molécula de HBr para halogenación, la molécula se convierte en
Br2 mediante el NBS.
3
Sustitución en alquenos por radicales libres:
orientación y
reactividad
Los grupos alquílicos de los
alquenos, por tanto, son sustituidos por halógenos exactamente del mismo modo
que los alcanos. Sin embargo, unido a estos grupos alquilo hay un sustituyente:
el doble enlace. Al igual que los alquilos afectan a la reactividad del doble
enlace, este último influye en la reactividad de los grupos alquílicos. Veamos
cuál es el efecto y cómo surge.
La halogenación de muchos
alquenos ha demostrado que: (a) los hidrógenos unidos a carbono con doble
enlace sufren muy poca sustitución, y (b) los hidrógenos conectados a carbonos adyacentes al doble enlace son
particularmente reactivos en la sustitución. El estudio de reacciones que
implican un ataque por otros radicales
libres, además de los átomos de halógeno, ha establecido como hidrógenos
vinílicos, son más difíciles de separar que los hidrógenos primarios
corrientes; los hidrógenos unidos a
carbonos adyacentes a un doble enlace, llamados alílicos, son aún más difíciles
de separar que los hidrógenos
terciarios.

Podemos ampliar ahora la
secuencia de reactividad de la sección 3.23.
Facilidad de separación
de alílico >
3º > 2º
> 1º > CH4
> vinílico
Átomos de hidrógeno
En los alquenos, la
sustitución procede por el mismo mecanismo que en los alcanos. Por ejemplo:
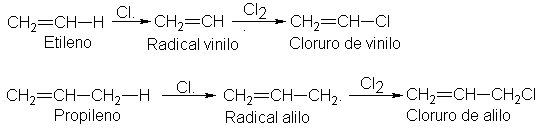
Es evidente que el radical
vinilo se forma muy lento, y el alilo, muy rápido. Ahora podemos ampliar la
secuencia de la sección 3.25.
Facilidad de formación
de alilo > 3º
> 2º >
1º > CH3.
> vinilo
Radicales libres.
·
¿Están de acuerdo estos
hallazgos con nuestra regla de que cuanto más estable es el radical, más
rápidamente se genera?
·
¿Es relativamente inestable
el radical vinilo de formación lenta?
·
¿Es relativamente estable
el radical alilo de generación rápida?
Las energías de disociación
de los enlaces indican que se necesita
una energía de 108 kcal para formar radicales vinilo desde un mol de etileno,
comparado con las 98 kcal necesarias para generar radicales etilo desde el
etano. En consecuencia, el radical vinilo contiene más energía y es menos
estable que un radical metilo, cuando se refiere al hidrocarburo del cual
deriva cada uno.
Por otra parte, las energías de disociación de enlaces
indican que sólo se necesitan 88 kcal para formar radicales alilo de propileno,
comparadas con las 92 kcal para los radicales t-butilo. En relación con el
hidrocarburo del cual se derivan, el radical alilo contiene menos energía y es
más estable que el t-butilo.
Podemos ampliar ahora la
secuencia de la sección 3.24; en relación con el hidrocarburo del cual se
generan, el orden de estabilidad de radicales libres es:
Estabilidad de radicales
libres alilo >
3º > 2º >
1º > CH3.
> vinilo
Entonces, el doble enlace
afecta de algún modo a la estabilidad de ciertos radicales libres; ejerce un
efecto similar sobre los radicales incipientes del estado de transición, con lo
que influye en la velocidad de su formación. Mediante estos efectos sobre la
velocidad de reacción, el doble enlace contribuye a determinar la orientación
de la sustitución en un alqueno por radicales libres y de las reactividades
relativas de alquenos diferentes. Así, el alqueno cíclico, ciclohexeno, se
broma casi exclusivamente en las posiciones alílicas, y reacciona más rápido
que el hidrocarburo saturado, ciclohexano, a pesar de un
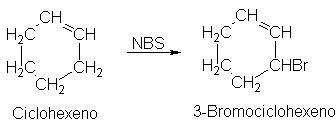
Factor de probabilidad de
12:4 favorable a un ataque en el compuesto saturado. (Problema: ¿Por qué 12:4?)
Sabemos que los radiales
libres no sólo se forman por extracción de hidrógeno, sino también por adición
a un doble enlace. Aquí, un doble enlace puede ayudar a determinar la
orientación y reactividad mediante su efecto sobre la estabilidad del radical
libre incipiente; sólo que ahora se trata de un segundo doble enlace, no del
que está sometido a la adición.
Ya dimos una posible
explicación para la baja estabilidad del radicales vinílicos. La unión de un
hidrógeno vinílico al carbono resulta del solapamiento de un orbital sp2 de
este último, en lugar de un sp3 de un carbono saturado, por lo que dicho enlace
es más corto y firme, y hay que invertir más energía para romperlo. En relación
con el hidrocarburo del cual procede, un radical vinílico es, por consiguiente,
relativamente inestable.
La elevada estabilidad de
los radicales alílicos puede explicarse, como veremos, por la teoría
estructural: específicamente por medio del concepto de resonancia. Sin embargo,
antes de ocuparnos de este concepto, estudiemos algunas características más de
los radicales alílicos que, al igual que sus estabilidades, son poco usuales.
4
Sustitución en alquenos por radicales libres:
transposición
alílica
Ya que utilizaremos el
radical alilo como introducción al concepto de conjugación y a la teoría de la
resonancia, examinaremos en detalle su estructura. Además del hecho de que:
·
el radical alilo es
especialmente estable, hay otros que deben justificarse por medio de una
estructura satisfactoria. Veamos cuáles son estos hechos.
·
La sustitución por
radicales libre en posiciones alílicas puede conducir a una transposición alílica.
Cuando se trata 1-octeno,
por ejemplo, con N-bromosuccinimida, no sólo se obtiene el 3-bromo-1-octeno
esperado, sino también y en
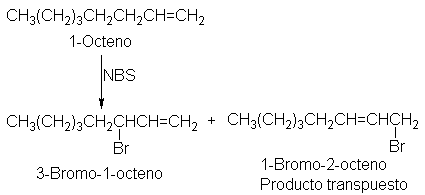
cantidad mayor
1-bromo-2-octeno (tanto Z como E). Es un hidrógeno alílico del C-3
lo que se extrae, pero en gran parte del producto aparece bromo en el C-1.
Cuando la estructura lo permite, esta transposición alílica sucede según un
esquema bien definido:
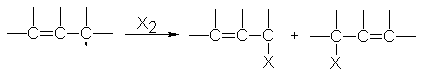
Podemos apreciar que el
radical alílico reacciona para dar dos productos distintos: uno, en el que el
halógeno se ha unido al carbono que perdió el hidrógeno, y otro, en el que el
halógeno se ha unido al carbono del otro extremo de la unidad de tres carbonos
el sistema alílico que representamos por C=C-C.
El examen de las estructuras
implicadas indica que tal transposición
no implica ninguna migración de átomos o grupos, solamente el doble enlace
aparece en una posición diferente de la que ocupaba en el reactivo.
5
Simetría del radical alilo
El radical alilo es una molécula simétrica.
Como ya hemos visto, un
doble enlace carbono-carbono es distinto de uno simple: es más corto y más fuerte; la rotación alrededor del enlace C-C
se halla impedida; los carbonos doblemente enlazado sujetan otros átomos
hidrógenos, por ejemplo mediante mediante enlaces más cortos y firmes.
Si el radical alilo
realmente tuviera la estructura clásica hasta ahora descrita,
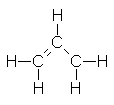
No sería simétrico en torno
al átomo de carbono central; es decir, los
extremos de la molécula serían diferentes entre sí. Contendría dos tipos de
enlaces carbono-carbono: uno simple, largo, y otro doble, corto.
Pues bien, un espectro RSE
(espectro de resonancia del espin electrónico) refleja la estructura de un radical libre, según puede observarse
en los hidrógenos de la molécula: entre otras cosas, indica cuántos tipos
diferentes de hidrógeno contiene el radical libre. Emite una señal por cada
hidrógeno, o por cada conjunto de hidrógenos equivalentes; esto es, cada conjunto
de hidrógenos en el mismo entorno .
Examinemos la estructura
clásica del radical alilo. Los dos hidrógenos vinílicos (Ha y Hb) en el carbono
terminal no serían equivalentes (diastereotópicos, en realidad), puesto que uno
es cis, y otro, trans con respecto al CH2. Los dos hidrógenos (Hc)
del CH2 serían equivalentes; debido a la rotación
rápida en torno al enlace simple carbono-carbono, ambos estarían en el mismo
entorno promedio. Finalmente, está el hidrógeno vinílico (Hd)sobre el carbono
central: diferente de todos los demás. En consecuencia, si el radical alilo
tuviera la estructura clásica, sería de esperar un espectro RSE correspondiente a cuatro tipos de
hidrógenos.
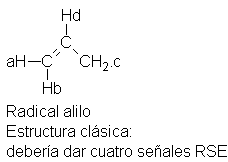
Sin embargo, el
espectro realmente medido solamente revela tres tipos de
hidrógenos.
Todo hidrógeno vinílico de
un extremo de la molécula tiene una contraparte idéntica en el otro extremo.
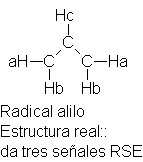
(Los dos hidrógenos
marcados Ha son equivalentes, y también lo son los marcados Hb.) Los dos extremos
de la molécula son equivalentes; los enlaces carbono-carbono son exactamente
del mismo tipo. El radical alilo es perfectamente simétrico en torno al carbono central.
Nuestra estructura clásica
del radical alilo es evidentemente insatisfactoria. Se requiere una estructura
que explique la notable estabilidad de este radical, la existencia de las
transposiciones alílicas y la simetría revelada por la RSE. Para conocer esta
estructura, debemos analizar la teoría de la resonancia.
6
Teoría de la resonancia
Será útil exponer primero
algunos de los principios generales del concepto de resonancia, y analizarlos
luego en función de un ejemplo específico: la estructura del radical alilo.
·
Siempre que se pueda
representar una molécula por dos o más
estructuras que sólo difieren en el ordenamiento de los electrones esto
es, mediante estructuras que presentan el mismo ordenamiento de los núcleos
atómicos-, hay resonancia. La molécula es un híbrido de todas esas estructuras,
y no puede representarse satisfactoriamente por ninguna de ellas. Se dice que
cada una de esas estructuras contribuye al híbrido.
·
Cuando las estructuras
contribuyentes tienen aproximadamente la misma estabilidad (el mismo contenido
energético), la resonancia es importante. La contribución de cada estructura al
híbrido depende de la estabilidad relativa de esa estructura: las estructuras
más estables dan mayor contribución.
·
El híbrido de resonancia es
más estable que cualquiera de las estructuras contribuyentes. Este aumento de
la estabilidad se denomina energía de resonancia. Cuanto más iguales sean las
estabilidades de las estructuras contribuyentes, mayor será la energía de
resonancia.
Solo puede haber resonancia
entre estructuras que contengan el mismo número de electrones impares. Sólo nos
interesa esta restricción al tratar di-radicales: moléculas que contienen dos
electrones no apareados. No puede haber resonancia entre una estructura de di-radical y otra con todos sus electrones apareados.
7
El radical alilo como híbrido de resonancia.
Así pues, desde el punto de
vista de la resonancia, el radical alilo es un
híbrido de resonancia de las dos estructuras, I y II.
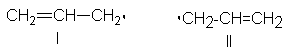
Esto significa simplemente
que el radical alilo no corresponde ni a I ni a II, sino más bien a una estructura intermediaria entre I
y II. Además, puesto que I y II son exactamente iguales, por lo que tienen la
misma estabilidad, el híbrido de resonancia está igualmente relacionado con I y
con II; o sea, se dice que I y II contribuyen al híbrido por igual.
Esto significa que la mitad
de las moléculas del radical alilo corresponden a I, y la otra mitad, a II, ni
tampoco que una molécula individual oscila entre I y II; todas ellas son
iguales, y cada una tiene una estructura intermediaria entre I y II.
Resulta útil una analogía
con híbridos biológicos propuesta por el
profesor G. W. Wheland (Universidad de Chicago). Cuando decimos que una
mula es un híbrido de caballo y burra, no estamos pensando que algunas mulas
son caballos y otras burros, ni tampoco que una mula sea durante un tiempo
caballo, y en otro momento, burra. Simplemente queremos indicar que una mula es
un animal relacionado tanto con el caballo como con el burro, y que puede
definirse convenientemente en función de esos animales que nos son familiares.
Es aún más adecuada una
analogía empleada por el profesor John
D. Roberts (Instituto Tecnológico de California). Un viajero medieval europeo
retorna a su hogar de un viaje a India y describe un rinoceronte como cruce
entre dragón y unicornio descripción bastante satisfactoria de un animal real
en función de dos animales familiares, pero totalmente imaginarios.
Debe entenderse que nuestra
imagen de dos estructuras para representar al radical alilo no implica que alguna
de ellas ( o las moléculas que cada una de ellas representa individualmente)
tiene existencia real. Las dos imágenes son necesarias para las limitaciones de
la inexactitud de nuestros métodos para presentar moléculas: hacemos dos
representaciones, porque una sencilla no sería suficiente. No es de extrañar
que ciertas moléculas no puedan representarse por una estructura del tipo que
hemos empleado; al contrario, lo sorprende es que las representaciones simples
de guiones y puntos utilizadas por los químicos orgánicos hayan funcionado tan
bien.
La teoría de la resonancia
nos dice, además, que el radical alilo no contiene un enlace simple carbono-carbono y otro doble (como se indica en
I y II), sino que tiene dos enlaces idénticos, cada uno intermediario entre uno
simple y uno doble. Este nuevo tipo de enlace híbrido ha sido descrito como un
enlace y medio. Se dice que posee la mitad de carácter de enlace simple, y la
mitad, de doble.
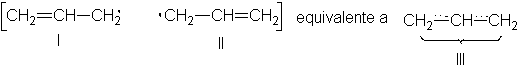
El electrón impar no se
encuentra en uno u otro carbono, sino que está deslocalizado, encontrándose
distribuido por igual sobre los dos carbonos
terminales. Podemos representar esta molécula híbrida simétrica como en
III, en donde las líneas de puntos
representan medios enlaces.
Hemos arribado, por cierto,
exactamente al tipo de estructura altamente simétrica indicada por el espectro
RSE del radical alilo.
La transposición alílica es
una consecuencia natural del carácter híbrido de un radical alílico. Los
carbonos terminales del sistema alílico de tres carbonos son exactamente
Equivalentes en el propio
radical, y muy similares en un radical alílico sustituido asimétricamente.
Cuando un halógeno reacciona con tal radical, puede ligarse a cualquiera de
estos carbonos terminales. Donde la estructura lo permite, como en el 1-octeno,
por ejemplo, esta unión queda demostrada a ambos extremos por la formación de
dos productos diferentes. En el caso del propio radical alilo no sustituido, se obtiene el mismo
producto, cualquiera que
Sea el extremo que recibe
al halógeno, por lo que no se observa
una transposición; sin embargo, tampoco cabe duda de que en este caso ambos
carbonos están sometidos al ataque.
8
Estabilidad del radical alilo
Otra consecuencia muy
importante de la teoría de la resonancia es la siguiente: como híbrido de
resonancia, el radical alilo es más estable (o sea, contiene menos energía) que
cualquiera de las estructuras contribuyentes. Esta estabilidad adicional de la
molécula se denomina energía de resonancia. Puesto que estas estructuras
contribuyentes específicas son exactamente equivalentes y, en consecuencia, de
igual estabilidad, esperamos que la estabilización debida a la resonancia sea grande.
¿Puede decirse lo grande
que es la energía de resonancia del radical alilo? Para conocer el valor
exacto, deberíamos comparar el radical alilo híbrido real con uno inexistente
de estructura I o II, algo que experimentalmente no es posible. Sin embargo,
podemos estimar la energía de resonancia comparando dos reacciones: la
disociación del propano para generar un radical n-propilo, y la disociación del
propeno para formar un radical alilo.
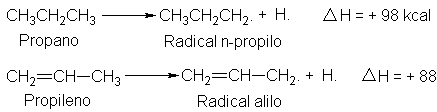
Propano, propileno y el
radical n-propilo pueden representarse de forma satisfactoria por una sola
estructura; por otra parte, el radical alilo es un híbrido de resonancia.
Observamos que la diferencia energética entre propileno y el radical alilo es
10 kcal/mol menor (98-88) que la diferencia energética entre propano y el
radical n-propilo; atribuimos la menor
energía de disociación enteramente a la estabilización por resonancia del
radical alilo y estimamos que la energía correspondiente es 10 kcal/mol.
9
Descripción orbital del radical alilo
Para tener un cuadro más
claro de lo que es un híbrido de resonancia y, en especial, para comprender
cómo surge la estabilización por resonancia-, consideremos los orbitales de
enlace del radical alilo.
Puesto que cada carbono
está unido a otros tres átomos, utiliza orbitales sp2. El
solapamiento de estos orbitales entre sí, y con los s de cinco átomos de
hidrógenos genera el esqueleto molecular indicado en la figura 10.1, con todos
los ángulos de enlace de 120º. Además, cada átomo de carbono tiene un orbital
p, que, como sabemos, consta de dos lóbulos iguales, uno por encima y el otro por
debajo del plano de los enlaces o, y que está ocupado por un solo electrón.
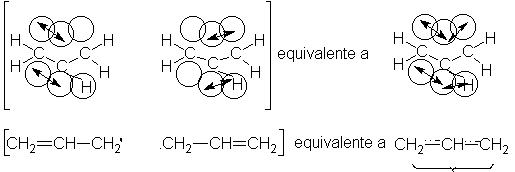
Como en el caso del
etileno, el orbital p de un carbono puede solapar al correspondiente de un átomo
de carbono adyacente, permitido así el apareamiento de electrones y la
formación de un enlace. De este modo obtenemos cualquiera de las dos
estructuras contribuyentes, I o II, en
las que el electrón no apareado ocupa
un orbital p del carbono restante. Pero el solapamiento no se limita a un par
de orbitales p, como es el caso del etileno; el orbital p del carbono central
solapa igualmente bien los orbitales p de los dos carbonos unidos a él. El
resultado es un par de nubes electrónicas n continuas, una por encima y otra
por debajo del plano de los átomos.
Debido a que el mismo
orbital no puede ocupar más de dos electrones
(principio de exclusión de Pauli), estas nubes p se componen de dos orbitales. Uno de los, que contiene dos electrones p, abarca los tres átomos de carbono; el otro,
que contiene el tercer electrón p (no apareado), está
distribuido por partes iguales entre los carbonos terminales.
El solapamiento de los
orbitales p en una y otra dirección, y la participación resultante de cada electrón
en dos estructuras. Estos dos métodos de representación, el dibujo de varias
estructuras resonantes y el de una nube electrónica, son sólo intentos
imprecisos para transmitir, por medio de dibujos, la idea de que un par de
electrones determinado puede servir para conectar más de dos núcleos. Esta
propiedad de los electrones p de participar en varios enlaces, esta deslocalización de electrones,
da origen a enlaces más fuertes y a una molécula más estable. Por esta razón,
frecuentemente se emplea el término energía de deslocalización, en vez de
energía de resonancia.
El enlace covalente debe su
fuerza a que un electrón es atraído con mayor intensidad por dos núcleos que
por uno. En forma análoga, un electrón es atraído más fuertemente por tres
núcleos que por dos.
El radical metilo puede no
ser enteramente plano, o sea, que la hibridación del carbono puede ser intermediaria entre sp2 y sp3. Para el
radical alilo, por el contrario y para muchos otros radicales libres-, la planaridad
es un requisito evidente que permite el solapamiento de orbitales p hacia la
estabilidad del radical.
Desde el punto de
vista de las estructuras convencionales
de los enlaces de valencia que utilizamos, es difícil representar una
estructura única que sea intermediaria entre dos, la I y la II. Por otra parte,
la descripción orbital brinda un cuadro bastante claro del radical alilo: la
densidad electrónica que enlaza al carbono central con cada uno de los otros es
intermediaria entre la de un enlace simple y la de un doble enlace.
Durante generaciones, los
químicos han utilizado la palabra conjugado para describir moléculas que
contienen dobles enlaces (o triples) y simples alternados: 1,3-butadieno, por
ejemplo, o (de manera especial) benceno. Se dio un nombre particular a los
compuestos con esta característica estructural, porque se observó que tenía
algunas propiedades peculiares en común.
Con la aparición de la
teoría de la resonancia en la década de 1930, se atribuyeron estas propiedades
peculiares de las moléculas conjugadas a la interacción de los orbitales n de
dos o más dobles enlaces: un solapamiento muy similar al que se acaba de
describir para el doble enlace de un radical alilo con el orbital p que
contiene el electrón impar. El significado de la palabra conjugación fue
ampliado para incluir la yuxtaposición similar de enlaces de cualquier tipo
tanto o como n o p-, que permite, una vez más, el solapamiento lateral.
En consecuencia, el radical
alilo es una molécula conjugada.
Interpretamos sus propiedades especiales, como lo haremos para otras
moléculas conjugadas, mediante el uso de la teoría de la resonancia. Se espera
que un doble enlace carbono-carbono tiene una función especial como
sustituyente cada vez que su ubicación en una molécula crea un sistema conjugado:
un sistema que, de acuerdo con nuestra interpretación, debe existir como
híbrido de resonancia.
10
Aplicación de la teoría de la resonancia
La gran utilidad y, por
consiguiente, el gran valor de la teoría de la resonancia consiste en que
mantiene el tipo simple, aunque impreciso, de representación estructural,
empleado hasta ahora en este libro. Es a menudo particularmente útil estimar la
estabilidad de una estructura basándose en su racionalidad. Si sólo puede
dibujarse una estructura razonable para una molécula, hay muchas probabilidades
de que esta estructura la describa adecuadamente.
El criterio de racionalidad
no es tan vago como puede parecer. Que una estructura particular nos parezca
razonable, significa que hemos encontrado antes un compuesto cuyas propiedades
se justifican por una estructura de ese tipo, que debe representar, en
consecuencia, un tipo de ordenamiento atómico y electrónico bastante estable.
Cada una de las estructuras contribuyentes del radical alilo, por ejemplo,
parece bastante razonable, porque conocemos compuestos que poseen
características de esta estructura, como los alquenos y los radicales libres.
Hay varios criterios que
podemos emplear para estimar estabilidades relativas y, en consecuencia, la
importancia relativa de estructuras contribuyentes. Uno de éstos, tiene que ver
con (a) la electronegatividad y la ubicación de carga.
Un método conveniente para
indicar la polaridad (carácter iónico) del enlace hidrógeno-cloro, por ejemplo,
es la representación del HCI como un híbrido de las estructuras I y II.
Juzgamos que II tiene estabilidad apreciable y su contribución es importante,
porque la carga negativa se encuentra sobre un átomo muy electronegativo: el
cloro.

Por otra parte, consideremos
que el metano está representado adecuadamente por la estructura única III.
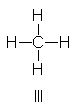
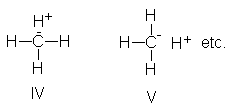
Aunque es posible dibujar
estructuras iónicas adicionales, con III o IV y V, consideraremos que son inestables, porque en ellas se
ubica una carga negativa sobre un
átomo de baja electronegatividad, el carbono. Supongamos que IV y V
tenga una contribución despreciable al híbrido, por lo que las ignoramos.
En secciones posteriores
emplearemos nuevos criterios para ayudarnos a estimar estabilidades de posibles
estructuras contribuyentes: (b) número de enlaces ; (c) dispersión de carga
;(d) octeto completo contra incompleto; (e) separación de cargas.
Finalmente, estudiaremos
ciertos casos para los cuales hay mucha evidencia longitudes de enlaces,
momentos dipolares, reactividad de que la descripción precisa de una molécula
determinada requiere la contribución de estructuras de un tipo que nos puede
parecer bastante irracional (Sec. 11 y 16); esto nos hace recordar que es muy
poco lo que sabemos acerca de la estructura molecular, por lo que debemos estar
preparados para modificar nuestra idea de lo que es razonable, para ajustarla a
la evidencia proporcionada por los hechos experimentales.
En la próxima sección
encontraremos estructuras de apariencia realmente extraña.
11
Estabilización por resonancia de los
radicales alquilo.
Hiperconjugación
Observemos en este punto
una extensión de la teoría de la resonancia que implica un tipo de conjugación,
a pesar de no implicar un doble enlace.
Las estabilidades relativas
de los radicales alquilo terciarios, secundarios y primarios se explican con el
mismo argumento empleado para justificar la estabilidad del radical alilo: la
deslocalización de los electrones, esta vez mediante el solapamiento entre el
orbital p, ocupado por el electrón impar, y un orbital o del grupo alquilo
(Fig. 10.2). Por medio de este solapamiento, los electrones pueden contribuir,
en cierto grado, a la unión de tres núcleos: dos carbonos y un hidrógeno. Este
tipo de deslocalización, que comprende orbitales o, se denomina hiper-conjugación.
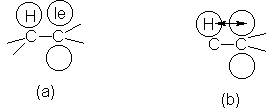
Desde el punto de vista de
la resonancia, el radical etilo, por ejemplo, es un híbrido, no sólo de la
estructura usual I, sino de tres adicionales, II, III y IV, donde los dos
carbonos están unidos por un doble enlace, y el electrón impar está retenido
por un hidrógeno.
Individualmente, cada una
de estas estructuras de resonancia sin enlace parece extraña, pero, en
conjunto, dan a entender que el enlace carbono-hidrógeno es algo menos que un
enlace simple, el enlace carbono-carbono
tiene algún carácter de doble enlace, y que el electrón impar es
acomodado parcialmente por los átomos de hidrógeno. La contribución de estas
estructuras inestables no es, de ninguna manera, de la importancia de las
estructuras equivalentes para el
radical alilo, por ejemplo, y que la estabilización resultante tampoco es tan
grande. Sin embargo, se estima que estabiliza al radical etilo hasta unas 6
kcal, en relación con el radical metilo, para el que la resonancia es
imposible.
Si ampliamos esta idea al
radical isopropilo, descubriremos que, en vez de tres estructuras de
hiperconjugación, ahora tenemos seis (dibújelas). El mayor número de
estructuras contribuyentes significa mayor deslocalización del electrón impar
y, por tanto, mayor estabilziación del radical. De acuerdo con lo dicho,
resulta que la energía de disociación de los enlaces de los hidrógenos
isopropílicos es sólo de 95 kcal, lo que indica una energía de resonancia de 9
kcal/mol (104-95).
Debería haber nueve
estructuras hiperconjugadas para el radical t-butilo (escríbalas).
Aquí encontramos una
energía de disociación de enlace de 92
kcal, lo que indica una estabilización por resonancia de 12 kcal/mol (104-92).
En resumen, las estabilidades
relativas de los radicales libres están determinadas por la deslocalización de
los electrones, que resulta del solapamiento de los orbitales p ocupados por el
electrón impar: solapamiento con una nube n de un doble enlace en el radical
alilo, o solapamiento con enlaces o en radicales alquilo.
Propuesta en 1939 por R. S.
Mulliken (Universidad de Chicago), la idea de la hiperconjugación ha levantado
mcuha controversia, al menos en algunas de sus aplicaciones. Se ha investigado
mucho, y se sigue investigando, en el intento de evaluar la importancia de los
efectos hiperconjugativos.
12
El catión alilo como híbrido de resonancia.
Volvamos a la química
heterolítica, y veamos cómo la afecta la presencia de un doble enlace en la
molécula del sustrato. Puesto que los carbocationes son intermediarios clave en
gran parte de la química heterolítica, comencemos con el examen del catión
alilo:

Antes de examinar los
hechos, veamos las predicciones que podemos hacer acerca de este carbocatión, utilizando
la teoría de la resonancia. Como siempre, examinaremos la estructura de la
molécula. Hemos dibujado el catión alilo como I, pero podríamos haberlo escrito
igualmente como II. Reconocemos de inmediato, ahora, que las estructuras I y II
cumplen con las condiciones de la resonancia: estructuras que sólo difieren en
la disposición electrónica.
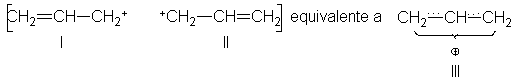
Según la teoría de la
resonancia, ni I ni II representan adecuadamente el catión, ya que se trata de un híbrido de I y II, cuya
estructura podríamos representar como III. Al ser I y II exactamente equivalentes y, en consecuencia, de la misma
estabilidad, contribuyen al híbrido por igual. Al igual que el radical alilo,
el catión alilo no contiene un enlace simple carbono-carbono ni uno doble:
tiene dos enlaces idénticos, cada uno intermediario entre un enlace simple y
uno doble. La carga positiva no se localiza en ninguno de los carbonos
terminales, sino que se atribuye sobre los dos.
Lo mismo que para el
radical alilo, podemos obtener un esquema más claro de esta molécula si
examinamos los orbitales enlazantes implicados. En ambas estructuras
contribuyentes hay un orbital p vacío en el carbono electrónicamente
deficiente. El solapamiento de este orbital p vacío con la nube n del doble
enlace resulta en la deslocalización de los electrones n: cada uno de estos dos
electrones ayuda a mantener juntos a los tres núcleos de carbono (Fig.3)
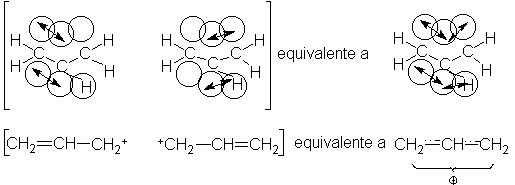
Pues bien, basados en la
estructura que hemos deducido, ¿qué predicciones podemos hacer en cuanto a las
propiedades del catión alilo? En primer lugar, y puesto que I y II son
exactamente equivalentes, esperamos que
la resonancia sea importante, dando origen a una estabilización considerable de
la molécula.
Esta predicción la
confirman los hechos, como se muestra en la tabla 1.3 (Sec. 1.14). La energía
de disociación heterolítica para el cloruro de alilo es de 173 kcal, 12 kcal
menos que para el cloruro de n-propilo, y aproximadamente la misma que para el
cloruro de isopropilo (170 kcal). Así, aunque la estructura I o II es
formalmente la de un catión primario, el catión alilo es casi tan estable como
uno secundario. Podemos ampliar ahora la secuencia de la sección 5.20.
alilo
Estabilidad de
carbocationes 3º
> 2º > 1º
> CH3+
En segundo lugar, esperamos
que el catión alilo sea simétrico en torno al carbono central.
De nuevo, los hechos
confirman esta aseveración. El catión alilo y los cationes alílicos sustituidos
en forma simétrica han sido preparados en condiciones fuertemente ácidas y
estudiados espectroscópicamente. El espectro infrarrojo de tal catión es
especialmente revelador: no aparecen dos bandas de absorción para el
estiramiento carbono-carbono, una para C-C y otra para C=C; en cambio, sólo
aparece una. Esta banda se encuentra en una frecuencia intermedia entre las
características de C-C y las de C=C, lo que indica dos enlaces C=C
equivalentes. La intensidad de esta banda se trata de la banda infrarroja más
intensa observada para compuestos orgánicos es indicativa de un sistema con una
carga positiva ubicada en ambos carbonos terminales.
13
Sustitución nucleofílica en sustratos alílicos: SN1.
Reactividad. Transposición alílica
Nuestros pronósticos sobre las
propiedades del catión alilo han sido correctos hasta aquí.
Veamos ahora lo que podemos
esperar de una reacción, donde los cationes alílicos son intermediarios: la
sustitución nucleofílica del tipo SN1.
Consideremos la solvólisis
del cloruro de alilo, por ejemplo,
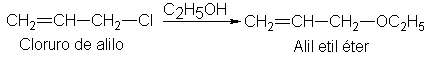
O la reacción del alcohol
alílico con un halogenuro de hidrógeno.
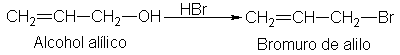
Como se desprende de los
nombres, son sustratos alílicos, puesto que el grupo saliente está ligado a un
carbono doble adyacente al carbono del doble enlace. ¿Cuál es el efecto de este
doble enlace sobre la reacción de estos sustratos?¿Se comportarán de manera
diferente que sus análogos saturados cloruro de n-propilo y alcohol
n-propílico, por ejemplo?
Supongamos por ahora que
estas reacciones siguen el mecanismo SN1. De acuerdo con éste, la etapa que
determina la velocidad es la heterólisis para generar un carbocatión, y, como
hemos visto, es la naturaleza de este carbocatión la que controla mayormente el
curso
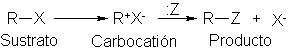
De la reación. En estos
casos, como los sustratos son alílicos, el catión intermediario será el
alílico. De lo que acabamos de ver acerca del catión alilo, ¿qué podemos
predecir acerca de estas reacciones SN1?
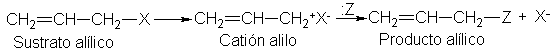
En SN1, la velocidad de la
formación del carbocatión es la que determina la velocidad total de la
reacción. Hasta ahora, hemos establecido que la velocidad con la que se forman
los carbocationes trátese de sustitución nucleofílica, eliminación o adición es
paralela a sus estabilidades. En la sección anterior, concluimos que el catión
alilo es tan estable como uno secundario. Por tanto, esperamos que los
sustratos alílicos reaccionen aproximadamente tan rápido por SN1 como los
sustratos secundarios.
Una vez más, estamos en lo
cierto. La solvólisis de sustratos alílicos
(cloruro o tosilato de alilo, por ejemplo) parece seguir un mecanismo
SN1 casi a la misma velocidad que la
reacción correspondiente de sustratos secundarios. (A veces, incluso, es algo
más rápida.)
Hemos visto que los
sustratos secundarios reaccionan mucho más velozmente por SN1 que los primarios
(Recuérdese: Es muy probable que todo o casi todo lo medido para la velocidad
de la solvólisis de sustratos primarios (Sec. 5.22) corresponde a una reacción
por SN2. En consecuencia, en una sustitución nucleofílica se forma el catión
alilo como el catión secundario quizás un millon de veces más rápido que su
análogo saturado, el catión n-propilo.
Velocidad de la
formación
alilo
De carbocationes 3º >
2º > 1º
> CH3+
La reactividad que hemos
estado analizando antes corresponde a sustratos que contienen el propio grupo
alilo simple, CH2=CH-CH2. La presencia de sustituyentes alquilo en cualquiera
de los extremos del sistema alílico aumenta aún más la reactividad.
Hagamos una predicción más:
esperamos que las reacciones SN1 de sustratos alílicos muestren transposiciones
alílicas. En el segundo paso del SN1, la combinación del carbocatión con el
nucleófilo debería ocurrir en cualquiera de los carbonos terminales del sistema
alílico, dando así, si la estructura lo permite, dos productos diferentes.
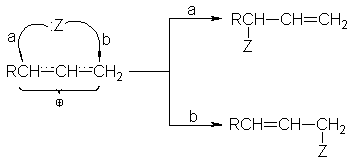
¿Cuáles son los hechos?
Consideremos la conversión de los cloruros alílicos isómeros IV y V en los
correspondientes etil éteres, VI y VII. En una solución concentrada de etóxido
de sodio en etanol, cada cloruro reacciona con una cinética de segundo orden;
IV da exclusivamente VI, y V da VII. Con una concentración elevada de un
nucleófilo fuerte, la reacción
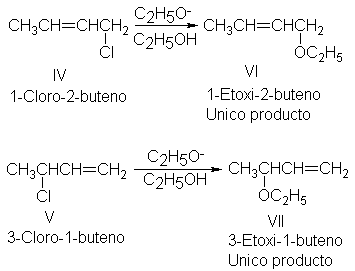
es directamente SN2; el
nucleófilo entrante se une al mismo átomo que ésta perdiendo el ión cloruro.
Si ahora se calientan los
mismos cloruros en etanol sin base añadida, cambia drásticamente el curso de la
reacción. Cualquiera que sea el sustrato alílico con que comencemos, se
encuentran ambos éteres en el producto. En condiciones solvolíticas, la
reacción cambia al mecanismo SN1; tal como anticipamos, hay transposición
alílica.
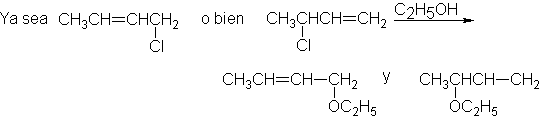
En el ejemplo presentado,
señalamos que los cloruros isómeros dan el mismo catión alílico híbrido. Si
este fuera exactamente el caso, esperaríamos obtener mezclas idénticas de
productos de ambos sustratos. Esto no es cierto en este caso, ni tampoco en la
mayoría de las transposiciones alílicas. Aun cuando ambos cloruros dan mezclas
de los mismos productos isómeros, la composición exacta de la mezcla las
proporciones de los dos productos difieren ligeramente y depende del cloruro
del cual se obtuvo.
Si lo anterior se piensa a
fondo, descubriremos algo que, al fin, no es de extrañar. Los cationes formados
por heterólisis no son idénticos por dos razones. Primera, hay evidencia de que
los intermediarios son pares iónicos. (En efecto, algunas de las pruebas más
convincentes y clásicas para la mediación de pares iónicos en la solvólisis
fueron aportadas por el estudio de tales sistemas alílicos.) Estos pares
iónicos, por supuesto, no serán idénticos; la estructura exactas de par iónico
la ubicación del ión cloruro dependerá del carbono que haya abandonado el ión
cloruro: C-1 o C-3.
Segunda, es probable que
los cationes alílicos se formen, como los secundarios, por ayuda nucleofílica.
Esto, también, debe depender del
sustrato, y la estructura de los dos cationes nucleofílicamente solvatados será
diferente. (Incluso puede ser que una fracción de la reacción ocurra mediante
un mecanismo SN2 completo.)
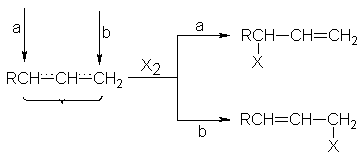
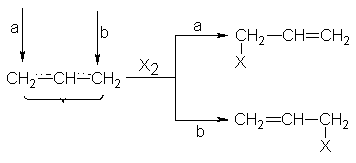
14
Estabilización de carbocationes:
efecto de resonancia.
En nuestra presentación de
los carbocationes, mencionamos que sus estabilidades relativas eran la
propiedad más importante para la comprensión de su química. Desde entonces,
hemos visto el notable paralelismo que existe entre la estabilidad de los
carbocationes y la de los estados de transición que llevan a su formación;
estados de transición de reacciones de muchos tipos diferentes.
La estabilización de un
carbocatión depende de la dispersión de
la carga, que, a su vez, proviene de la eficacia con que el carbono deficiente
en electrones puede adquirir electrones de otra parte de la molécula. Una
manera de que suceda esto es por el efecto inductivo de un sustituyente: la
liberación de electrones actuando a través de la armazón molecular o del
espacio, con debilitamiento gradual al aumentar la distancia entre el
sustituyente y el centro de la reacción. (La atracción electrónica tiene, por
supuesto, el efecto contrario: intensifica la carga y desestabiliza el
carbocatión.)
Hemos encontrado una
segunda manera de dispersar una carga: el efecto de resonancia (o efecto
conjugativo), debido al solapamiento de ciertos orbitales. A la inversa del
efecto inductivo, el de resonancia no varía gradualmente en su fuerza con la
distancia. Es un efecto de todo o nada, que depende de una relación específica
entre los átomos que interactúan: la relación que hemos llamado conjugación.
Hay dos
características implicadas en la
determinación de la estabilidad de la estructura de un carbocatión: el orbital
p, aun cuando está formalmente vacío y la planaridad en torno al carbono
deficiente en electrones. Veremos ahora cómo resulta esto: en un carbocatión
conjugado, el orbital p vacío está disponible para el solapamiento que
proporciona electrones al carbono deficiente, y la planaridad permite que este
solapamiento sea geométricamente posible.
El carbono electrónicamente
deficiente puede conjugarse con átomos o grupos distintos de un simple doble
enlace carbono-carbono: muy en especial
con un grupo arilo en lo que se denominan cationes bencílicos. El orbital p
vacío puede solapar a otros que no sean n: electrones no compartidos de un
átomo adecuadamente ubicado; incluso, quizás, orbitales o de los enlaces
carbono-hidrógeno. En todos estos casos, la deslocalización de electrones y la
dispersión de carga estabilizan el carbocatión.
En algunas reacciones no es
una carga positiva, sino una negativa la que se desarrolla.
Encontraremos que los
compuestos aniónicos formados más fácilmente, más estables y más importantes
son conjugados, y deben su estabilidad, y también su importancia, a la
dispersión de su carga mediante la resonancia.
La deslocalización de
electrones por resonancia es uno de los factores polares más poderosos que
afecta a la estabilidad de moléculas cargadas, positivas o negativas, y como
tal desempeña un papel muy importante en la determinación de la orientación y
la reactividad en una amplia variedad de reacciones orgánicas, e incluso en el
propio curso de la reacción.
15
Estabilización de carbocationes:
función de los pares no compartidos
Observemos un ejemplo de
conjugación de un c arbono deficiente en electrones con un átomo que posee
pares de electrones no compartidos.
El cloroéter, cloruro de
metoximetilo, sufre solvólisis (evidentemente por el mecanismo SN1) con una
rapidez superior es más de 104 veces la del cloruro de metilo incluso más
rápida que la de los cloruros de alquilo simples de cualquier clase. ¿Cómo se
puede justificar este aumento enorme de la velocidad causado por el grupo
CH3O-?
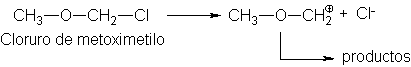
Como siempre en las
reacciones SN1, observemos la estructura del carbocatión intermediario. El
carbono deficiente en electrones tiene unido un átomo de oxígeno. El oxígeno es
electronegativo y, como un halógeno, debe ejercer un efecto inductivo de
atracción electrónica un efecto que tiende a desestabilizar un carbocatión
(Sec. 5.25). No obstante, en estye caso tenemos una prueba de estabilización,
poderosa, si sigue siendo válido el paralelismo entre velocidad de formación y
estabilidad.
Este paralelismo es válido:
las mediciones demuestran que el catión metoximetilo ers 76 kcal/ mol más
estable que el catión metilo es más estable incluso que el t-butilo. ¿Puede
entonces el oxígeno liberar electrones? La respuesta es sí, por su efecto de resonancia.
Aunque electronegativo, el
oxígeno del grupo CH3O es básico; tiene pares de electrones no compartidos que
tiende a compartir, adquiriendo así una carga positiva. Así como el agua acepta
un protón para generar un ión hidronio (oxonio),
![]()
También los alcoholes y
éteres los aceptan, como sabemos, para formar iones oxonio sustituidos.
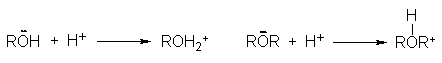
Los efectos sobre la estabilidad
de carbocationes de un oxígeno adecuadamente ubicado en este caso, y en otros
tipos de reacciones pueden explicarse admitiendo que el oxígeno comparte más de
un par de electrones con un carbono electrónicamente deficiente y puede
acomodar una carga positiva. Fundamentalmente, lo que está implicado aquí es la
basicidad del oxígeno.
Con estos antecedentes,
volvamos a la estructura del catión metoximetilo. Hemos escrito que su
estructura es I, pero igualmente pudimos presentarla como II. Una vez más, se
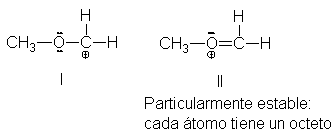
Cumplen las condiciones
para la resonancia: dos estructuras que sólo difieren en la disposición
electrónica.
De las dos estructuras,
suponemos que II es mucho más estable que I, pues cada átomo tiene un efecto un
octeto electrónico completo (excepto el hidrógeno). Al compartir dos pares de
electrones con el carbono lso electrones que precisaba para completar su
octeto. La estructura II es mucho más estable por sí sola que I, de manera que
representa con bastante exactitud la estructura del catión. Así, este catión
apenas merece ser considerado como un carbocatión; antes bien es un ión oxonio.
Este ión oxonio generado del cloruro de metoximetilo es simple, por lo que se
forma con una velocidad inmensamente mayor.
(Comparemos, por ejemplo,
las estructuras del H3O+ y
el CH3+. No se trata de explicar aquí qué átomo, oxígeno o
carbono, acomoda mejor una carga positiva; es una cuestión de octetos completos
o incompletos.)
Se trata de un sistema
conjugado: el doble enlace nace del solapamiento de un orbital p vacío de un
carbono con uno p lleno del oxígeno. El carbono electrónicamente deficiente de
un carbocatión puede conjugarse con un par no compartido de átomos distintos
que el oxígeno: nitrógeno, azufre, incluso halógeno. También en estos casos la
estabilización del
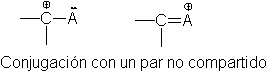
Carbocatión resultante
puede tener efectos especulares sobre la velocidad, no sólo de la heterólisis,
sino de las reacciones de muchos otros tipos.
Por tanto, en el ión oxonio,
la liberación de electrones por medio del efecto de resonancia es mucho más
poderosa que la atracción de electrones mediante el efecto inductivo, y
controla la reactividad. Veamos ahora una situación en la que los factores
están más equilibrados. Volvamos a la formación de un carbocatión en una
reacción de un tipo diferente, la adición electrofílica, y examinemos el efecto
de un elemento distinto, el cloro.
Como ejemplo, consideremos
la adición de yoduro de hidrógeno al cloruro de vinilo y consideremos la
reactividad y la orientación en esta reacción. Los hechos son los que siguen:
la adición es más lenta que al etilo, y se obtiene 1-cloro-1-yodoetano.
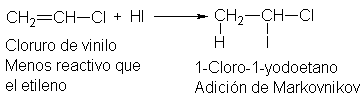
Examinemos primero el
aspecto de la reactividad. Vimos que la adición electrofílica es un proceso de
dos pasos. El primero, lento, es la generación de un carbocatión cuya
estabilidad determina la velocidad de su formación y, por tanto, la de la
propia adición. La adición al etileno da el catión etilo, III; la adición al
cloruro de vinilo da el
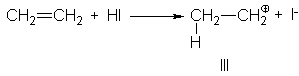
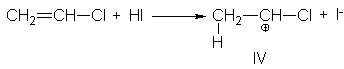
Catión 1-cloroetilo, IV. El
efecto inductivo de atracción de electrones del cloro intensifica la carga
positiva en IV, hace estable el catión y causa una reacción más lenta.
Hasta aquí todo va bien.
Pero veamos la orientación de esta reacción.
Sabemos (Sección 8.12) que la orientación en la adición electrofílica está
determinada por cual de dos carbocationes posibles se forma en el primer paso;
de nuevo, se genera más velozmente el carbocatión más estable. La adición al
cloruro de vinilo podría implicar el catión IV o el V.
El producto que se obtiene
realmente, 1-cloro-1-yodoetano, demuestra que se forma
Preferentemente IV, por lo
que quizá sea el más estable. No obstante, en IV la carga positiva está ubicada
sobre C-1, la posición más cercana al cloro, donde suponemos que efecto
inductivo será máximo y más desestabilizante. ¿Cómo justificar esta orientación
desordenada?
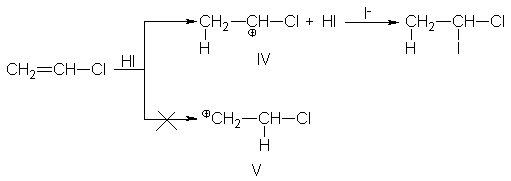
La respuesta se encuentra
en la conjugación: conjugación entre el carbono electrónicamente deficiente y un
par de electrones no compartido del cloro. El catión que realmente se forma no
está representado de forma adecuada por la estructura IV; es un híbrido de IV
con la estructura VI, donde el carbono y el cloro están unidos por un doble
enlace, y el cloro
Lleva la carga
positiva. Al igual que el ión oxonio ya
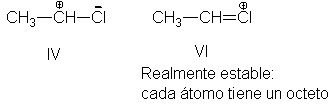
mencionado, la estructura VI es relativamente estable, porque en ella cada
átomo (excepto hidrógeno) tiene un octeto completo. En el catión V alternativo,
que tiene carga positiva sobre C-2, no hay conjugación comparable, ni ninguna
contribución de una estructura como la VI. En consecuencia, en la medida que la
estructura VI contribuye al híbrido, hace que el catión que tiene la carga
sobre C-1 sea el más estable.
Mediante su efecto inductivo,
el cloro tiende a atraer electrones, desestabilizando así al carbocatión
intermediario. Este efecto se percibe en los dos carbonos, pero más
intensamente en C-1. Por medio del efecto de resonancia, el cloro tiende a
liberar electrones, estabilizando así el carbocatión intermediario. Esta
liberación electrónica sólo es efectiva en C-1.
El efecto inductivo es más
fuerte que el de resonancia y ocasiona una atracción electrónica neta, con la
consiguiente desactivación de la molécula en relación con el etileno no
sustituido. El efecto de resonancia tiende a oponerse al inductivo para la
generación del
Catión con carga en C-1,
por lo que éste queda menos desestabilizado que el catión alternativo.
La reactividad queda así
controlada por el efecto inductivo más fuerte, y la orientación, por el de
resonancia que, aunque más débil, es más selectivo.
encontraremos la misma acción recíproca de los efectos inductivo y
de la resonancia en la determinación de la orientación y reactividad en una
reacción la sustitución aromática electrofílica, que superficialmente parece
ser bien distinta de ésta, pero que es básicamente muy similar.)
Para justificar la
estereoquímica de al adición de los halógenos a alquenos se ha propuesto un ión
halogenonio intermediario: se considera que el halógeno comparte dos pares de
electrones y adquiere una carga positiva. A pesar de la elevada
electronegatividad del halógeno, tal intermediario es más estable que un catión
abierto, de acuerdo con la evidencia estereoquímica. En el efecto de resonancia
descrito, la estabilidad relativa, igual que en la formación de los iones halogenonio cíclicos, es una cuestión de
octetos completos o incompletos, y no de la electronegatividad del átomo
portador de la carga positiva.
16
Estabilización por resonancia de cationes alquilo:
hiperconjugación
Observemos otro lugar donde
un carbono electrónicamente deficiente de un carbocatión puede obtener
electrones: los enlaces carbono-hidrógeno. Se ha propuesto que el orbital p
vacío de un carbocatión, como el de un radical libre, puede solapar orbitales o
de los grupos alquilo a los que se encuentra unido.
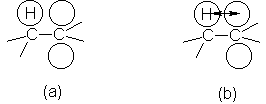
Como se ha visto al atraer
los radicales libres, este tipo de solapamiento permite que electrones
individuales contribuyan a enlazar entre sí tres núcleos: dos carbonos y un
hidrógeno. Conocemos como hiperconjugación esta clase de deslocalización.
Desde el punto de vista de
la resonancia, se describiría el catión etilo, por ejemplo, como un híbrido no
sólo de la estructura I, sino de las tres estructuras II, III y IV, en las
cuales ambos carbonos están unidos mediante un doble enlace, y la carga
positiva es transportada
Por un hidrógeno. Según el
grado en que el enlace carbono-carbono haya adquirido carácter de doble enlace,
el carbono electrónicamente deficiente adquiere electrones, dispersándose la
carga positiva entre los tres hidrógenos.
Con el aumento de la
ramificación del carbocatión, se incrementa la dispersión de carga y la estabilización:
la dispersión en el catión isopropilo es sobre seis hidrógenos, y en el
t-butilo, sobre nueve.
Anteriormente describimos la liberación de electrones por
grupos alquilo como un efecto inductivo; aquí estamos viendo que es un efecto
de resonancia. A pesar del enorme trabajo llevado a cabo en relación con este
problema, no se ha aclarado la importancia relativa de estos dos factores.
Frecuentemente, se encuentran reunidos como el efecto
inductivo-hiperconjugativo de los grupos alquilo. En esta obra nos referimos a
menudo al efecto inductivo de los grupos alquilo, pero debe entenderse que
puede incluir una contribución de la hiperconjugación.
17
Sustitución nucleofílica en sustratos alílicos. SN2
Volvamos al comportamiento
de los sustratos alílicos en la sustitución nucleofílica. Vimos que son casi
tan reactivos en eacciones SN1 como los sustratos secundarios saturados. Esto
se atribuyó a la dispersión, por medio de la resonancia, de la carga positiva
en desarrollo en el estado de transición del paso determinante de la velocidad.
Ahora bien, en la
sustitución nucleofílica por SN2 se establece que los sustratos
alílicos son casi tan reactivos como los sustratos primarios saturados. (A menudo, los sustratos alílicos son algo
más reactivos hasta unas 40 veces.) Lo anterior es comprensible: el factor
principal que gobierna la reactividad SN2 es el impedimento estérico, y el
grupo alilo es casi tan voluminoso como uno primario no ramificado.
En cierto sentido, el grupo
alilo tiene lo mejor de dos mundos: una capacidad para la dispersión de carga
comparable a la de un grupo secundario, pero sin la voluminosidad, que le
impediría el ataque nucleofílico directo.
18
Sustitución nucleofílica en sustratos vinílicos
Continuemos con el examen
del efecto del doble enlace sobre la sustitución nucleofílica, y observemos
sustratos en los que el grupo saliente está ligado a uno de los carbonos del doble enlace; esto es,
sustratos vinílicos.
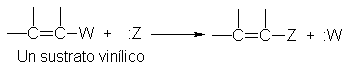
Hemos visto que un halogenuro de alquilo se detecta
apropiadamente por la precipitación de halogenuro de plata insoluble al
calentamiento con nitrato de plata alcohólico.
Esta reacción es un ejemplo
de sustitución nucleofílica una solvólisis, donde el ión plata presta
ayuda, extrayendo un ión halogenuro.
Esta reacción es instantanea con bromuros terciarios, alílicos y bencílcios, y
tarda unos cinco minutos con bromuros primarios y secundarios.
Los halogenuros vinílicos
(o de arilo), Sin embargo, no dan halogenuro de plata en estas condiciones.
Puede calentarse bromuro de vinilo con AgNO3 alcohólico durante días, sin que
se detecte AgBr. Frente a la sustitución nucleofílica en general, los
halogenuros vinílcios son muchísimo menos reactivos que sus análogos saturados.
Normalmente no se les utiliza en el conjunto de síntesis basadas en reacciones de sustitución
nucleofílica de halogenuros de alquilo.
¿Cómo podemos explicar esta
baja reactividad de los halogenuros vinílicos? Fundamental para la comprensión
de estos compuestos es que poseen un enlace carbono-halógeno para el cloruro de
vinilo es de 207 kcal, en comparación con las 191 kcal para el cloruro de
etilo, y 227 kcal, para el cloruro de metilo. Los valores para fluoruros,
bromuros y yoduros presentan diferencias similares. Para romper el enlace
carbono-halógeno es un halogenuro de vinilo, se necesitan entre 16 y 18 kcal
más de energía que en el halogenuro de etilo correspondiente. A excepción del
enlace en los halogenuros de metilo, el enlace carbono-halógeno es el más
fuerte que hemos encontrado hasta ahora.
Dos son los factores
propuestos que intervienen en la fuerza inusual del enlace vinilo-halógeno: (a)
resulta del solapamiento con un orbital sp2 del carbono, en lugar
del sp3 de un carbono saturado ; (b) tiene algún carácter de doble
enlace, debido a la resonancia. Posteriormente, trataremos este asunto con más
detalle , por ahora sólo diremos que es probable que operen los dos factores.
También es probable que
estos dos mecanismos factores refuercen la unión entre un carbono doblemente
enlazado y el oxígeno de grupos salientes como los tosilatos. En cualquier
caso, los sustratos vinílicos que contienen otros grupos salientes de uso común
comparten esta baja reactividad de los halogenuros vinílicos. Así, el tosilato
de (E)-2-buten-2-ilo reacciona con metanol acuoso sólo a una millonésima de la
velocidad del tosilato de sec-butilo.
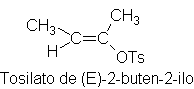
En consecuencia, la unión
vinilo-halógeno es muy fuerte. Ahora bien, si la sustitución nucleofílica
ocurre por SN2 SN1, el paso determinante de la velocidad implica la ruptura del
enlace carbono-halógeno. El enlace más fuerte en los halogenuros de vinilo es
más difícil de romper, de manera que la reacción es más lenta. Teniendo
presente lo anterior, examinemos estas reacciones más a fondo.
19
Sustitución nucleofílica en sustratos vinílicos: cationes vinílicos
Como veremos pronto,
ciertos sustratos vinílicos especiales sufren sustitución nucleofílica por SN1,
aunque, evidentemente, no por SN2. Por tanto, consideremos primero la
sustitución del tipo SN1. Tales reacciones de sustratos vinílicos implicarían,
claro está, cationes vinílicos.
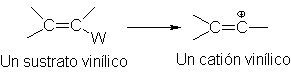
La reactividad
extremadamente reducida de los halogenuros vinílicos por SN1 indica que los
cationes vinílicos se forman con mucha lentitud a partir de estos sustratos. En
consideración al paralelismo observado entre la estabilidad carbocatiónica y la
velocidad de su generación, nos preguntamos: ¿son relativamente inestables los
cationes vinílicos?
Acabamos de ver la prueba
de que es así. Si se necesitan 16-18 kcal más de energía para formar cationes
vinílicos a partir de halogenuros vinílicos que para formar cationes etílicos a
partir de halogenuros de etilo, entonces los cationes vinílicos son, por
definición, menos estables que los cationes de etilo en relación con el
halogenuro del cual se genera cada uno.
De la misma manera, los
cationes vinílicos son más estables que los metílicos en 16 a 20 kcal. Así
pues, podemos ampliar nuestra secuencia de la sección 10.12.
alilo
Estabilidad de carbocationes 3º >
2º > 1º >
vinilo > CH3+
(Mientras no descubramos lo
contrario, ha de resaltarse que la posición del catión vinilo en esta secuencia
sólo es apropiada para su estabilidad en relación con sustratos para
heterólisis.
¿Cómo justificamos esta
baja estabilidad del catión vinilo? Recordemos la forma en que planteamos este
tipo de problema: la medida de la estabilidad de un carbocatión es,
sencillamente, la cantidad de energía necesaria para generarlo del sustrato
elegido. Esta cantidad de energía varía de catión en catión. Hasta aquí, es un
hecho.
Ahora comienza el proceso
teórico. Tratamos de explicar estas variaciones para hallar el esquema más
amplio al cual se ajustan. Buscamos
factores que justifiquen estas variaciones y que, al mismo tiempo, sean
congruentes con el resto de la teoría estructural. En nuestro análisis de la
estabilidad carbocatiónica hemos centrado nuestra atención en las diferencias
entre los propios cationes. Por ejemplo, la subida energética del cloruro de
t-butilo al catión t-butilo es menor que la del cloruro de isopropilo al catión
correspondiente, porque decimos que el nivel de energía del catión t-butilo es
rebajado por dispersión de carga. No hay ningún factor particular que haga muy
diferentes al cloruro de t-butilo y al de isopropilo; en cambio, sí hay uno que
distingue a los cationes entre sí: la
dispersión de carga. Pues bien, este
análisis ha dado buen resultado hasta ahora.
Sin embargo, en la
generación de cationes vinílicos la sustitución es diferente. Primero, como
veremos en la sección 11.9, es posible generar cationes vinilo sin mayores
dificultades, por una reacción distinta de la heterólisis. Segundo, es claro
que los halogenuros de vinilo difieren considerablemente de los halogenuros de
alquilo saturados de cualquier clase. A pesar de las diferencias en estabilidad
entre cationes 1º, 2º y 3º, los enlaces
carbono-halógeno de los halogenuros de vinilo son significativamente más
cortos: esto concuerda con la imagen de una unión más compacta y fuerte, debida
al solapamiento de un orbital sp2 de un carbono y al carácter de doble enlace
parcial.
Por consiguiente, nuestra
interpretación es como sigue: a partir
de halogenuros orgánicos, la subida energética al catión vinilo es superior que
a la del catión etilo, no porque están operando factores que elevan el nivel
energético del catión vinilo, sino porque algunos rebajan el nivel energético
del halogenuro de vinilo.
Esto es un ejemplo de un
principio químico fundamental. Al interpretar diferencias en reactividades,
tendemos a buscar factores que estabilizan más un estado de transición que otro
que refleja generalmente el carácter de producto que posee, rebajando así la Eact;
este enfoque opera bien la mayoría de las veces. Sin embargo, debemos estar
siempre alerta frente a factores que pudieran estabilizar más un reactivo que
otro, con lo que Eact se incrementa.
Lo dicho hasta ahora está
relacionado con la dificultad para generar cationes vinílicos por heterólisis.
No debe llamarnos la atención que esta dificultad sea un desafío para el
químico orgánico y haya surgido cationes vinílicos como intermediarios
accesibles, con propiedades extraordinarias, en trabajos realizados
principalmente alrededor de 1970. Han estado involucrados en estas
investigaciones muchas personas de numerosos países, entre ellas Michael Hanack
(Universidad de Tubinga), Zvi Rappaport (Universidad Hebrea de Jerusalén),
Giorgio Modena (Universidad de Padua) y Peter Stang (Universidad de Utah).
Es fácil generar cationes
vinílicos por solvólisis del tipo SN1, si se cumplen dos condiciones: (a) el
grupo saliente tiene que ser extremadamente bueno, y (b) el grupo vinílico debe
tener sustituyentes que liberen electrones.
Para este propósito, se
suelen utilizar el super grupo saliente trifluorometanosulfonato, OSO2CF3,
conocido como triflato.
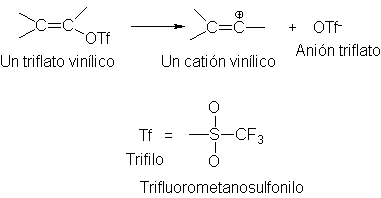
Los átomos de flúor,
poderosos extractores de electrones (por dispersión de la carga negativa),
contribuyen a estabilizar el anión triflato, CF3SO2O-, y
convierten al ácido correspondiente, CF3SO2OH, en uno de
los ácidos de Lowry-Bronsted más fuertes que se conocen mucho más fuerte que
los ácidos comunes H2SO4 y HCIO4-.
Correspondientemente, el anión triflato es una base muy débil, y uno de los
mejores grupos salientes de la química orgánica. En relación con la solvólisis,
se ha detectado que los triflatos de alquilo saturados son de 10 000 a 100 000
veces más reactivos que los tosilatos correspondientes, y hasta mil millones de
veces más reactivos que los cloruros o bromuros.
Los sustituyentes
liberadores de electrones en la parte vinílica son muy a menudo grupos arilo,
pero bastan grupos alquilo para permitir la reacción por SN1. Por ejemplo:
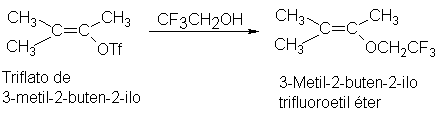
No podemos dedicar más
tiempo a tratar las propiedades de los cationes vinílicos; sin embargo, vale la
pena mencionar que, al igual que los cationes alquilo saturados, tienen una
química rica y variada: pueden generarse de diversos tipos de saturados en
distintos tipos de reacciones; conducir
tanto a la eliminación como a la sustitución, y pueden transponerse.
Quizá la lección más conveniente que podemos aprender
de lo anterior no sea la química de los cationes vinílicos por importante que
parezca. Lo interesante aquí es que vemos resuelto un problema de modo lógico
reconociendo los fundamentos estudiados: el valor que para un proceso
heterolítico tiene (a) un grupo saliente bueno débilmente básico y (b) la
liberación de electrones en el catión que se está generando.
Lo que acabamos de analizar
es la sustitución nucleofílica unimolecular SN1. Habíamos dicho antes que la
reacción por SN2 no parece ocurrir con sustratos vinílicos. Esta reactividad
más baja ha sido atribuida al impedimento estérico para el ataque por atrás del
nucleófilo: un impedimento debido a la nube n.
Si se utilizan nucleófilos
fuertes y si el sustrato vinílico contiene grupos que atraen electrones, puede
ocurrir una sustitución bimolecular, que, sin embargo, no es SN2. Por ser
difícil la reacción por la vía SN2, sigue un curso más aliviado: un mecanismo
de un tipo diferente, que no implica, significativamente, la ruptura del enlace
al grupo saliente en el paso determinante de la velocidad. También es
significativo el hecho de que este mecanismo se parezca mucho al tipo de
sustitución nucleofílica característico de los halogenuros de arilo otra clase
de sustratos con enlaces carbono-halógeno inusualmente fuertes.
20
Dienos: estructura y propiedades
En este capítulo se ha mencionado
el efecto del doble enlace, actuando como sustituyente, sobre ciertas
reacciones que ocurren en otra parte de la molécula: sustitución por radicales
libres y nucleofílica. Veamos ahora su efecto sobre la química del alquenos;
esto es, el efecto de un doble enlace en la química de otro enlace doble en la
misma molécula: en su formación y en las reacciones que sufre.
Haremos esto principalmente
mediante el estudio de dienos, que son alquenos con dos dobles enlaces. Lo que
expondremos es igualmente aplicable a compuestos con más de dos dobles enlaces.
El enlace doble de un dieno tiene las mismas propiedades que el de los alquenos
ya estudiados. Sin embargo, en ciertos dienos, estas propiedades son
modificadas por la presencia de un segundo doble enlace. Fijaremos nuestra
atención en modificaciones.
Los dienos se dividen en
tres clases, según la disposición de sus dobles enlaces. Cuando estos enlaces
dobles alternan con enlaces simples son conjugados. Los dobles enlaces
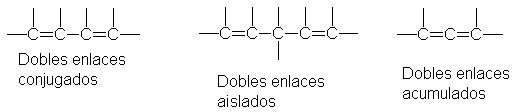
Separados por más de uno
simple se llama aislados. Aquellos dobles enlaces que comparten un carbono son
acumulados, y los compuestos se denominan alenos. Ejemplos:
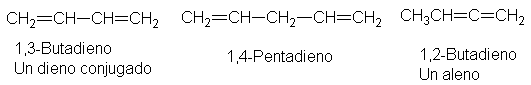
Las propiedades químicas de
un dieno dependen de esta disposición de sus dobles enlaces. Los dobles enlaces
aislados ejercen escasas influencia recíproca, por lo que cada uno de ellos
reacciona como si fuera el único doble enlace de la molécula. Salvo por el mayor consumo de reactivos, las
propiedades químicas de los dienos no conjugados son idénticas a las de los
alquenos simples. Los alenos tienen cada vez mayor interés para los químicos
orgánicos, aunque apenas tendemos tiempo para dedicarnos a ellos.
Concentraremos nuestra
atención en los dienos conjugados, que difieren de los alquenos simples en
cuatro aspectos: (a) son más estables; (b) son los productos preferidos en las
eliminaciones; (c) sufren adición 1,4 tanto electrofílica como por radicales
libres, y (d) son más reactivos en la
adición de radicales libres.
21
Estabilidad de dienos conjugados.
Si prestamos atención a la
tabla 8.1 (Sec. 8.3), observaremos que los valores de hidrogenación de los
alquenos con estructuras similares son notablemente constantes. Para los
alquenos monosustituidos (RCH=CH2), los valores son muy cercanos a 30 kcal/mol;
para los disustituidos (R2C=CH2 o RCH=CHR), 28 kcal/mol, y para los
trisustituidos (R2C=CHR), 27 kcal/mol. Para un compuesto con más de un doble
enlace, esperaríamos un calor de hidrogenación igual a la suma de los calores
de hidrogenación de los dobles enlaces individuales.
Para los dienos no
conjugados, esta propiedad es aditiva, como puede apreciarse en la tabla
10.1 para el 1,4-pentadieno y el
1,5-hexadieno, por ejemplo, cuyos valores son cercanos a 2 x 30 kcal, o 60
kcal/mol.
Sin embargo, para los dienos conjugados, los
valores medidos son ligeramente más bajos de lo esperado. Para el 1,3-butadieno
debería ser 2 x 30, o 60 kcal/mol: el valor real, 57 kcal, es 3 kcal menor. Del
mismo modo, los valores para el 1,3-pentadieno y el 2,3-dimetil-1,3-butadieno
también son inferiores a lo esperado en 2 4 kcal.

¿Qué nos revelan estos
calores de hidrogenación acerca de los dienos conjugados?
Empleando los argumentos de
la sección 8.4, comparemos, por ejemplo, el 1,3-pentadieno (calor de hidrogenación,
54 kcal) y el 1,4-pentadieno (calor de hidrogenación, 61 kcal).
Ambos consumen dos moles de
hidrógeno y dan el mismo producto, el n-pentadieno. Si el 1,3-pentadieno libera
menor energía que el 1,4-pentadieno, sólo puede significar que contiene menos
energía; es decir, el 1,3-pentadieno conjugado es más estable que el
1,4-pentadieno no conjugado.
En las tres secciones
siguientes veremos cómo se recurre a dos factores distintos para explicar las
estabilidades relativas de los dienos conjugados y de los alquenos simples (a)
deslocalización de electrones n, (b) fortalecimiento de enlaces o por el cambio
de hibridación del carbono.
22
Resonancia en dienos conjugados
Estudiaremos ahora los cuatro
átomos principales de carbono de cualquier sistema diénico conjugado. De
ordinario, escribimos los enlaces C(1)-C(2) y C(3)-C(4) como dobles, y el
C(2)-C(3), como simple.
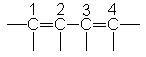
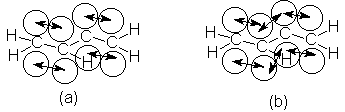
Esto correspondería a una
representación orbital de la molécula (véase Fig. 10.5a), donde se forman
enlaces n por solapamiento de los orbitales p de C-1 y C-2, y C-3 y C-4.
 En el radical alilo pudimos apreciar que la
resonancia era el resultado del solapamiento del orbital p de un átomo de
carbono con orbitales p a ambos lados. De igual forma, podemos suponer que
podría haber cierto solapamiento entre los orbitales p de C-2 y C-3, como se
indica en la figura 10.5b. La deslocalización resultante de los electrones p estabiliza la molécula: cada par de
electrones atrae y es atraído por no sólo dos núcleos de carbono, sino cuatro.
En el radical alilo pudimos apreciar que la
resonancia era el resultado del solapamiento del orbital p de un átomo de
carbono con orbitales p a ambos lados. De igual forma, podemos suponer que
podría haber cierto solapamiento entre los orbitales p de C-2 y C-3, como se
indica en la figura 10.5b. La deslocalización resultante de los electrones p estabiliza la molécula: cada par de
electrones atrae y es atraído por no sólo dos núcleos de carbono, sino cuatro.
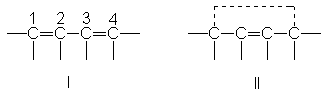 Empleando el lenguaje de las estructuras
convencionales de los enlaces de valencia, describimos un dieno conjugado como
híbrido de resonancia de I y II. La línea de puntos en II representa un enlace
formal y significa simplemente que un electrón del C-1 y otro del C-4 tienen
espines opuestos, ó que están apareados.
Empleando el lenguaje de las estructuras
convencionales de los enlaces de valencia, describimos un dieno conjugado como
híbrido de resonancia de I y II. La línea de puntos en II representa un enlace
formal y significa simplemente que un electrón del C-1 y otro del C-4 tienen
espines opuestos, ó que están apareados.
De acuerdo con el grado de
contribución de II a la estructura híbrida, da cierto carácter de doble enlace
al C(2)-C(3) y cierto carácter de enlace sencillo al C(1)-C(2) y al C(3)-C(4);
es más, hace más estable la molécula de lo que permite suponer I (la estructura
contribuyente más estable).
La formación de un enlace
libera energía y estabiliza un sistema; la igualdad de los demás factores,
cuanto mayor es el número de enlaces, mayor es la estabilidad de la estructura.
La consideración del número de enlaces es uno de los criterios (Sec. 10.10) que
puede usarse para calcular la estabilidad relativa y, por consiguiente, la
importancia relativa de una estructura contribuyente. Basados en esto, se
supone que II con 10 enlaces (no cuenta el enlace formal) es menos estable que
I con II enlaces. La energía de resonancia para tal híbrido de estructuras no
equivalentes debe ser menor que la de uno constituido por estructuras
equivalentes. La estructura de un dieno conjugado debe parecer más a I que a
II, puesto que I, más estable, contribuye más al híbrido.
De acuerdo con el carácter
de doble enlace parcial, el enlace C(2)-C(3) del 1,3-butadieno tiene una
longitud de 1.48 A, en comparación con 1.53 A de un enlace simple puro. La
energía de resonancia de un dieno conjugado es sólo de 2 a 4 kcal/mol, en
comparación con 10 kcal/mol del radical alilo.
23
Resonancia en alquenos. Hiperconjugación
Los calores de
hidrogenación mostraban que los
alquenos no sólo se estabilizan por conjugación, sino también por la presencia
de grupos alquilo: cuanto mayor es el número de lo que están unidos a los
carbonos del doble enlace, más estable es el alqueno. Para tomar el ejemplo más
sencillo, el calor de hidrogenación del propileno es 2.7 kcal menor que el del
etileno, lo que significa (en relación con el alcano correspondiente) que el
primero es más estable en 2.7 kcal que el etileno.
La estabilización por grupos
alquilo se ha atribuido al mismo factor fundamental que la estabilización por
un segundo doble enlace: la deslocalización de electrones, esta vez por
solapamiento entre un orbital n y uno o del grupo alquilo. Por medio de este
solapamiento, los electrones individuales pueden contribuir, hasta cierto
grado, a la unión de cuatro núcleos. Se denomina hiperconjugación a la
deslocalización de este tipo que implica enlaces.
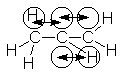
Utilizando la terminología
de la resonancia, dicha hiperconjugación se representa por la contribución de
estructuras como II. (La línea de puntos de II representa un enlace formal,
para indicar que los electrones de ambos átomos están apareados.)
Individualmente, una estructura como la II es realmente extraña, al no existir
un enlace real entre hidrógeno y carbono. Sin embargo, es una forma aproximada
de indicar que el enlace carbono-hidrógeno es algo menos que un enlace simple;
que el enlace C(2)-C(3) tiene algún carácter de doble enlace, y que el
C(1)-C(2) tiene algo de enlace simple.
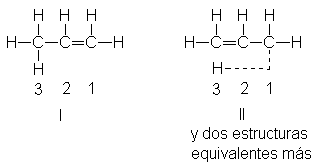
En relación con el carácter
de doble enlace parcial, el enlace simple carbono-carbono del propileno tiene
una longitud de 1.50 A, en comparación con 1.53 A de un enlace simple puro.
Cuanto mayor es el número
de grupos alquilo unidos a los carbonos del doble enlace, mayor es el número de
estructuras contribuyentes como la II, mayor es la deslocalización electrónica
y más estable es el alqueno.
La hiperconjugación del
tipo que acabamos de describir se llama hiperconjugación de sacrificio, pues en
las estructuras como II hay un enlace verdadero menos que en I. Por el
contrario, el tipo de hiperconjugación que vimos para los radicales libres y
los iones carbonio no implica ningún sacrificio de enlace y se llama
hiperconjugación isovalente.
24
Estabilidad de dienos y alquenos:
una interpretación alternativa
Hemos visto que la longitud
del enlace carbono-hidrógeno decrece a medida que avanzamos en la serie etano,
etileno, acetileno, lo que se atribuyó a cambios en la hibridación del carbono.
(Tabla 10.2.) A medida que disminuye el carácter p del orbital enlazante,
decrece el tamaño del orbital, por lo que el enlace se acorta.
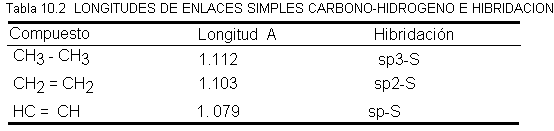
La longitud del enlace
simple carbono-carbono también disminuye en la serie análoga etano, propileno, propino
(Tabla 10.3); podemos observar que estas diferencias son mayores

Para los enlaces
carbono-hidrógeno. En este caso, el acortamiento de las uniones se atribuye a
la hiperconjugación estudiada en la sección 23.
Algunos químicos,
especialmente M. J. S. Dewar (Universidad de Texas), han dicho que no es
necesario recurrir a la hiperconjugación en moléculas como éstas, y que los
cambios en la longitud del enlace C-C, al igual que en el C-H, se deben
principalmente a cambios en la hibridación del carbono.
Dewar señaló también que
tal acortamiento de enlaces va acompañado de un aumento proporcional de las
energías de enlaces (E); es decir, que un acortamiento de un enlace estabiliza
la molécula. El cambio de hibridación afecta más a las longitudes de enlace y,
por tanto, a la estabilidad molecular cuando se ven implicados enlaces
carbono-carbono que cuando involucra
enlaces carbono-hidrógeno. Un sustituyente alquílico estabiliza un alqueno, en
relación con el alcano correspondiente, porque la hibridación sp2 fortalece más
un enlace carbono-carbono que uno carbono-hidrógeno.
De forma análoga, la
estabilidad especial de los dienos conjugados se atribuye aque la hibridación
sp2-sp2 acorta el enlace C(2)-C(3) (1.48 A), fortaleciéndolo, y no a la deslocalización
de electrones.
No cabe duda que operan
ambos factores: la deslocalización de electrones n y el cambio en los enlaces
o. ¿Cuál es la importancia relativa de cada uno? Bien puede suceder que la
respuesta sea que ambos son importantes.
Dewar no ha dudado de la
importancia de la deslocalización de electrones n en moléculas como el radical
alilo, para el cual, evidentemente, no es aceptable ninguna estructura única.
Considera, sin embargo, que la estabilidad del enlace o es más importante de lo
que suele reconocer. También, acepta que tiene un papel más importante la
hiperconjugación isovalente en radicales libres y carbocationes que la
hiperconjugación de sacrificio que acabamos de estudiar.
25
Facilidad de formación de dienos conjugados:
orientación de la eliminación
La mayor estabilidad de los
dienos conjugados se refleja en su mayor facilidad de formación. Son, cuando es
posible, los productos preferidos en las reacciones de eliminación. Ejemplo:
El dieno más importante,
1,3-butadieno (utilizado para la obtención de sustitutos de la goma, Sec.
10.30), se sintetiza industrialmente en cantidades considerables mediante el
cracking de hidrocarburos:
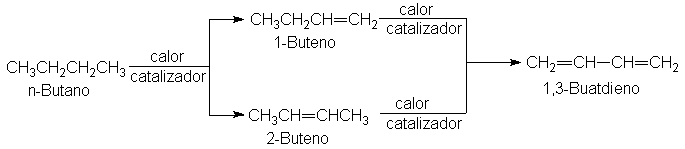
26
Adición electrofílica a dienos conjugados. Adición 1,4
Cuando el 1,4-pentadieno se
trata con bromo en condiciones que favorecen la formación del dihalogenuro
(¿cuáles son?), se obtiene 4,5-dibromo-1-penteno, el producto esperado. La
adición de más bromo produce el 1,2,4,5-tetrabromopentano. Este es el
comportamiento
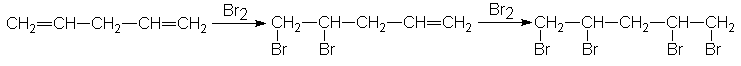
Típico de los dienos con
dobles enlaces aislados: los dobles enlaces reaccionan independientemente, como
si estuvieran en moléculas diferentes.
Cuando se trata el 1,3-butadieno
con bromo en condiciones similares, no sólo se obtiene el 3,4-dibromo-1-buteno
esperado, sino también el 1,4-dibromo-2-buteno. El tratamiento con HCI da
3-cloro-1-buteno y también 1-cloro-2-buteno. La hidrogenación genera 1-buteno y
2-buteno.
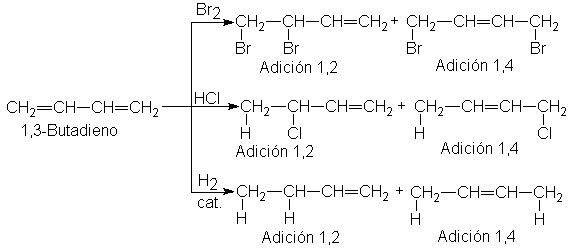
El estudio de muchos dienos
conjugados y de muchos reactivos demuestra que tal comportamiento es típico: en
las adiciones a dienos conjugados, un reactivo puede unirse no sólo a un par de
carbonos adyacentes (adición 1,2), sino también a los carbonos de los dos
extremos del sistema conjugado (adición 1,4). Con frecuencia, el producto de la
adición 1,4 es el principal.
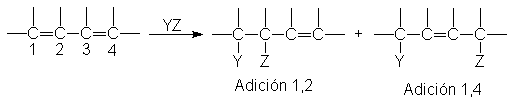
¿Cómo podemos explicar la
obtención de estos productos y, en particular, la ocurrencia de la adición 1,4?
Hemos visto que la adición electrofílica es un proceso de dos etapas, y que la
primera de ellas genera el carbocatión más estable. Apliquemos este principio a
la adición del HCI al 2,4-hexadieno, por ejemplo, que da 4-cloro-2-hexeno y
2-cloro-3-hexeno:
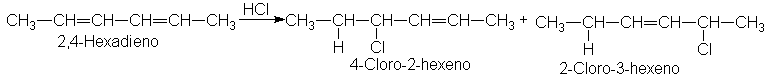
Estos productos revelan que
el hidrógeno se agrega al C-2 para dar el carbocatión I, en vez de al C-3 para
formar el carbocatión II:
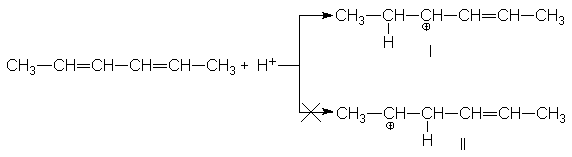
Puesto que I y II son
cationes secundarios, ¿cómo podemos explicar esta preferencia? La respuesta,
por supuesto, se encuentra en la estructura de I: no es simplemente un catión
secundario, sino también un catión alílico, debido a que el carbono portador de
la carga positiva está unido a un carbono con doble enlace. Por tanto, es un
híbrido de resonancia:
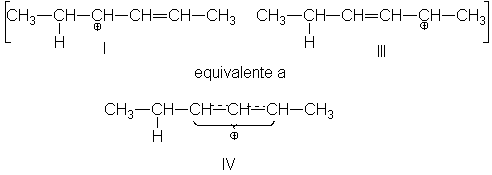
Como catión secundario y
alílico, I es más estable que II, y es el intermediario catiónico preferido.
Los productos obtenidos de
la adición a dienos conjugados siempre concuerdan con la formación del
carbocatión intermediario más estable: un catión alílico. Esto requiere que el
primer paso sea la adición a uno de los extremos del sistema conjugado. Ahora,
en el segundo

Paso, el catión IV se
combina con el ión cloruro para dar el producto. El ión cloruro puede unirse a cualquiera
de los extremos del sistema alílico, dando así el producto 1,2 ó 1,4.
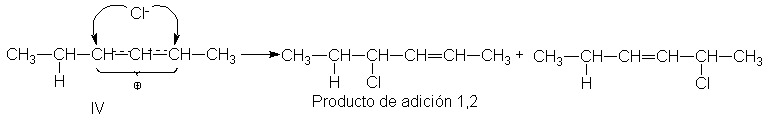
Al igual que en la
transposición alílica (Sec. 10.13), vemos que la adición 1,4 es una consecuencia natural de la naturaleza
híbrida del catión alílico intermediario.
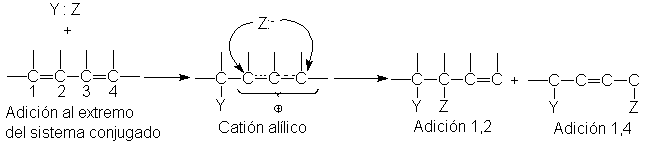
La naturaleza híbrida del
catión alílico gobierna así etapas de la adición electrofílica a dienos
conjugados: la primera, por la estabilización del catión; la segunda,
permitiendo la unión a cualquiera de dos átomos de carbono.
27
Adición 1,2 contra 1,4. Velocidad contra equilibrio
AL observar las cantidades
relativas que se obtienen de los productos de adición 1,2 y 1,4, surge un
principio muy importante.
La adición de HBr al
1,3-butadieno da los productos 1,2 y 1,4; las proporciones en que se obtienen
se ven marcadamente afectadas por la temperatura a la que se efectúa la
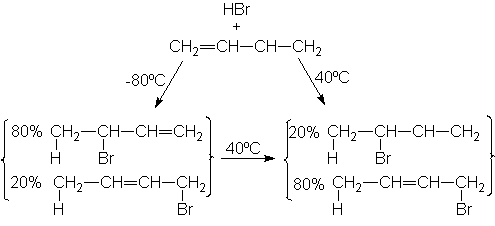
Reacción. A temperatura
baja (-80 ºC) da una mezcla con el 20% del producto 1,4 y el 80% del 1,2; a temperatura
más elevada (40 ºC) se obtiene una mezcla de composición bien diferente, con el
80% del producto 1,4 y el 20% del 1,2. A temperaturas intermedias se obtienen
mezclas de composición también intermedia. Aunque ambos isómeros son estables a
temperatura baja, el calentamiento prolongado del producto 1,4 o del 1,2 genera
la misma mezcla. ¿Cómo se interpretan estas observaciones?
El hecho de que el
calentamiento de cualquiera de los dos compuestos de la misma mezcla indica que
ésta es el resultado de un equilibrio entre ambas sustancias. El hecho de que
el compuesto 1,4 predomine en dicho equilibrio, prueba que es el más estable de
los dos.
Que se forme más producto
1,2 que 1,4 a –80 ºC, significa que el primero se genera más rápidamente que el
segundo; puesto que ninguno de ellos se modifica a –80ºC, las cantidades en que
se aíslan indican las proporciones en que se formaron inicialmente. A medida
que aumenta la temperatura de la reacción, las proporciones iniciales de los
productos pueden permanecer iguales, pero se acelera la conversión de los
productos iniciales en la mezcla de equilibrio.
Las proporciones de los
productos efectivamente aislados en la adición a baja temperatura están
determinadas por el equilibrio entre ambos isómeros.
Examinemos más
detenidamente la adición 1,2 y 1,4 trazando una curva de energía potencial para
las reacciones implicadas (Fig. 7). El primer carbocatión generado reacciona y
da más rápidamente el producto 1,2 que el 1,4; en consecuencia, la energía de
activación que conduce al compuesto 1,2 debe ser menor que la que lleva al
producto 1,4.
Esto lo representamos por
el máximo menor que va del ión al compuesto 1,2. Hay más colisiones con energía
suficiente para remontar éste que el máximo mayor, por lo que el producto 1,2
se forma más rápidamente que 1,4. En cambio, el compuesto 1,4 es más estable
que 1,2, por lo que debemos trazar su mínimo
a un nivel más bajo que el de la sustancia 1,2.
Como sabemos (Sec.13), los
halogenuros de alilo pueden sufrir heterólisis; es decir, se pueden ionizar.
Ahora, la ionización de cualquiera de los dos derivados bromados genera el
mismo carbocatión: la manera más probable y sencilla de que los productos 1,2 y
1,4 alcancen el equilibrio es por medio de este catión.
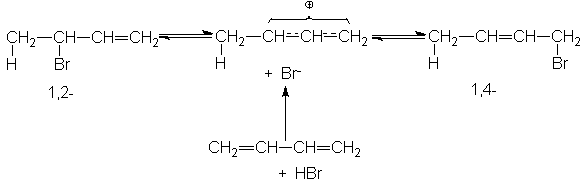
La ionización de los
bromuros implica remontar estos máximos de potencial de vuelta al carbocatión;
pero el ión está separado del compuesto 1,4 por un pico más alto que el del
compuesto 1,2, por lo que el primero se ionizará más lentamente que el segundo.
El equilibrio se alcanza cuando las velocidades de las reacciones opuestas son
iguales. El producto 1,2 es de formación más rápida, pero su ionización también
lo es; el producto 1,4 se genera lentamente, pero su ionización es aún más
lenta: una vez formado, este compuesto
tiende a persistir. A temperaturas lo suficientemente elevadas como para
alcanzar el equilibrio es decir, lo suficientemente altas como para que la
ionización sea significativamente rápida, predomina el compuesto 1,4, más
estable.
Nos hemos tratado de
explicar el hecho de que el producto 1,2 se forma más velozmente que 1,4 ni de
que éste sea más estable que aquél (aunque observamos que esto concuerda con
nuestra generalización de que los alquenos disustituidos son más estables que
los monosustituidos). Hemos aceptado estos hechos y demostrado lo que
significan desde el punto de vista energético. Se han observado relaciones
similares para otros dienos y reactivos.
Estos hechos ilustran dos
aspectos importantes. Primero, debemos ser cautelosos al interpretar la
composición de productos en función de la velocidad de reacción; debemos estar
seguros de que uno de ellos no se convierte en el otro después de su formación.
Segundo, el producto más
estable no tiene por qué formarse siempre más velozmente. Basados en muchas
pruebas, hemos deducido que por lo general cuanto más estable es un carbocatión
o radical libre, más rápido se forma; el estudio de los estados de transición
para estas reacciones ha demostrado que esto es posible. Sin embargo, no debemos
aplicar este principio a otras reacciones, a menos que las pruebas así lo
confirmen.
28
Adición de radicales libres a dienos conjugados: orientación
Al igual que otros
alquenos, los dienos conjugados sufren la adición de reactivos electrofílicos y
de radicales libres.En este último caso, los dienos conjugados presentan dos
aspectos especiales: sufren tanto la adición 1,4, como la 1,2 y son mucho más
reactivos que los alquenos ordinarios. Podemos explicar estas dos propiedades
orientación y reactividad examinando la estructura del radical libre
intermedio.
Como ejemplo, tomamos la
adición de BrCCI3 al 1,3-butadieno en presencia de un peróxido. Ya
sabemos que el peróxido se descompone
(paso 1) para generar un radical libre, el cual extrae bromo del BrCCI3
(paso 2) para generar un radical CCI3.
El radical CCI3
que resulta se agrega al butadieno
(paso 3). Los productos obtenidos demuestran que esta adición es a uno
de lso extrermos del sistema conjugado. ¿Por qué? En la adición de radicales
libres a dienos conjugados parece evidente que la orientación (como también la
reactividad) está controlada por la estabilidad del radical que se está
formando, y no por factores polares. En consecuencia, el CCI3 se añade a donde
lo hace porque de esta manera se genera un radical libre alílico estabilizado
por resonancia.
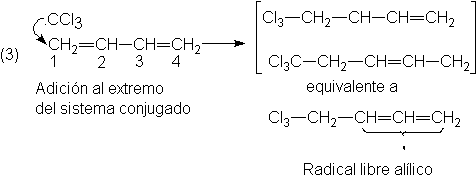
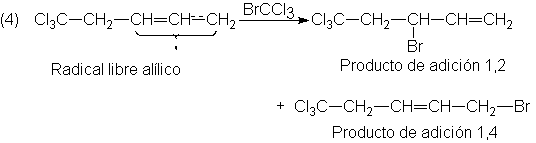 Luego, el radical alílico quita bromo de una
molécula de BrCCI3 (paso 4) para completar la adición y al proceder
así genera un nuevo radical CCI3 que puede continuar la cadena. En
el paso (4), el bromo puede unirse al C-2 o al C-4 para dar el producto 1,2 ó
1,4.
Luego, el radical alílico quita bromo de una
molécula de BrCCI3 (paso 4) para completar la adición y al proceder
así genera un nuevo radical CCI3 que puede continuar la cadena. En
el paso (4), el bromo puede unirse al C-2 o al C-4 para dar el producto 1,2 ó
1,4.
29
Adición de radicales libres a dienos conjugados: reactividad
Si se hace reaccionar BrCCI3
con una mezcla a partes iguales de 1,3-butadieno y un alqueno simple como
el 1-octeno, la adición sucede exclusivamente en el 1,3-butadieno. El radical
CCI3, evidentemente, se agrega mucho más velozmente al dieno
conjugado que al alqueno simple. Han sido observados resultados similares en
muchas reacciones de radicales.
¿Cómo puede explicarse la excepcional reactividad de los dienos conjugados? En nuestro estudio de la halogenación de los alquenos simples, vimos que tanto la orientación como la reactividad relativa estaban relacionadas con la estabilidad del radical libre formado en la primera etapa. Basados en esto, podemos suponer que la adición a un dieno conjugado que genera un radical libre alílico estable será más rápida que la adición a un alqueno simple. Hay indicios de que los factores polares son de escasa importancia en este caso.
Por otra parte, acabamos de
ver que los dienos conjugados son más
estables que los alquenos simples. Considerando esto, podríamos esperar que la
adición a dienos conjugados fuera más lenta que a alquenos simples.
La velocidad relativa de
cada reacción depende principalmente de sus valores de la Eact la
estabilización del radical libre alilo incipiente rebaja el nivel energético
del estado de transición y la estabilización del dieno disminuye la energía de
los reactivos. El que la Eact neta para la adición a un alqueno
simple sea mayor o menor depende de cuál se estabilice más (véase Fig. 8).
Los dienos conjugados con
más reactivos que los alquenos simples. Luego, en el presente caso y en la
mayoría de los casos que involucran alquenos y radicales libres, o alquenos y
carbocationes son más importantes los factores que estabilizan el estado de
transición, parece no ser válido para la adición electrofílica a dienos conjugados).
30
Polimerización de dienos por radicales libres.
Caucho y sustituidos del caucho
Al igual que los etileno
sustituidos, los dienos conjugados también pueden polimerizarse con radicales
libres. Por ejemplo, del 1,3-butadieno se obtiene un polímero cuya estructura
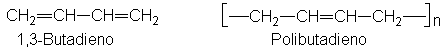
Indica que sucede
predominantemente la adición 1,4:
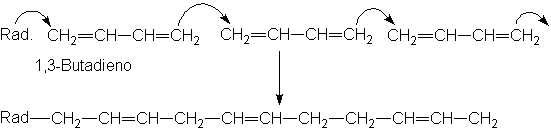
Este polímero difiere de
los obtenidos de alquenos simples en un aspecto muy importante: cada unidad aún
contiene un doble enlace.
El caucho natural tiene una
estructura muy semejante a la de estos polidienos sintéticos.
Podemos considerarlo un
polímero del dieno conjugado 2-metil-1,3-butadieno, o isopreno.
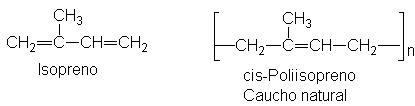
Los dobles enlaces de la
molécula del caucho son de gran importancia porque permiten la vulcanización
aparentemente proporcionando hidrógenos alílicos reactivos; es decir, la
formación de puentes de azufre cadenas diferentes. Estos enlaces cruzados
endurecen y dan mayor resistencia al caucho, y eliminan la pegajosidad del caucho
no tratado.

La polimerización de dienos
para obtener susitutos del caucho fue la precursora de la desarrollada
industria actual plásticos. El policloropreno (Neoprén, Duprén) fue el primer
sustituto del caucho de éxito comercial en Estados Unidos.
En parte, las propiedades
de los sustitutos del caucho como las de otros polímeros están determinadas por
la naturaleza de los grupos sustituyentes. El policloropreno, por ejemplo, es inferior
al caucho natural en algunas de sus propiedades, pero superior en su
resistencia a aceites, gasolina y otros disolventes orgánicos.
También puede obtenerse
artificialmente polímeros del isopreno; contienen la misma cadena no saturada y
el mismo sustituyente (el grupo CH3) que el caucho natural. Pero el
poli-isopreno obtenido por el proceso de radicales libres que presentamos, era
en relación con las propiedades que realmente importan muy inferior al natural.
Su estereoquímica era distinta: el caucho natural tiene la configuración cis en
(casi) todos sus dobles enlaces; el material artificial era una mezcla de cis y
trans. No pudo lograrse un caucho sintético verdadero hasta 1955; lo que se
necesitaba era un tipo de catalizador totalmente nuevo, además de un mecanismo
de polimerización enteramente diferente. Con ellos se hizo posible la
polimerización estereo-selectiva del isopreno, obteniéndose un material
virtualmente idéntico al caucho natural: el cis-1,4-poliisopreno.
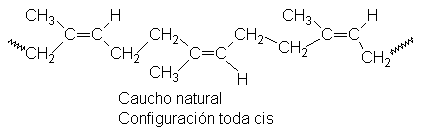
31
Isopreno y la regla isoprénica
La unidad isoprénica es uno
de los bloques constructivos favoritos de la naturaleza. No sólo aparece en el
caucho, sino también en una amplia variedad de compuestos que se aislan de
fuentes vegetales y animales. Por ejemplo, casi todos los terpenos (se
encuentran en los aceites esenciales de muchas plantas) tienen esqueletos
carbonados construidos con unidades de isopreno unidas entre sí de un modo
regular de de pies a cabeza. El
reconocimiento de este hecho la llamada regla isoprénica ha sido de gran ayuda
en la comprensión de las estructuras de los terpenos.
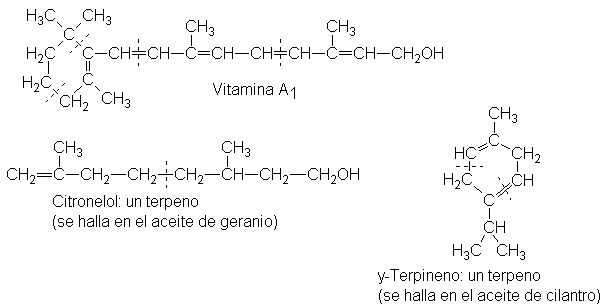
Un área fascinante de
investigación que une la química orgánica con la biología es el estudio de la biogénesis
de productos naturales: la serie de reacciones que forman un compuesto en los
sistemas vivos, plantas o animales. Aparentemente, todas las unidades de
isopreno de la naturaleza tienen su
origen en una misma sustancia: el pirofosfato de isopentenilo.
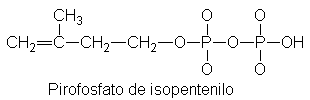
Trabajos realizados desde
1950, han demostrado que compuestos aparentemente tan diferente del caucho como
el colesterol se construyen, paso a
paso, a partir de unidades isoprénicas.
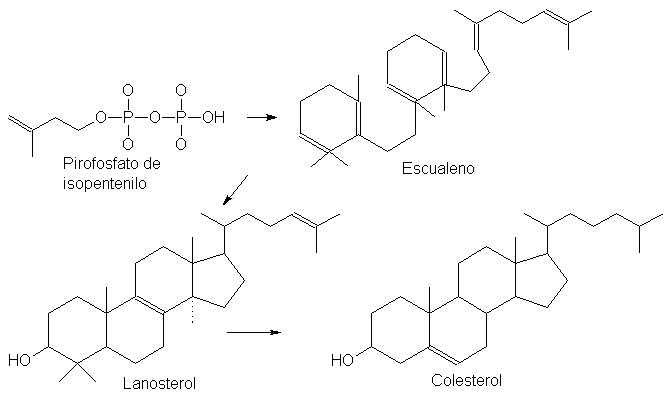
32
Análisis de dienos
Los dienos responden a las
pruebas de caracterización en la misma forma que los alquenos: decoloran el
bromo en tetracloruro de carbono sin desprendimiento de bromuro de hidrógeno, y
decoloran el permanganato diluído, neutro y frío; no son oxidados por el
anhídrido crómico. Son, sin embargo, más insaturados que los alquenos,
propiedad que puede detectarse por determinación de sus fórmulas moleculares (CnH2n-2)
y por una hidrogenación cuantitativa (se consumen dos moles de hidrógeno por mol
de hidrocarburo).
La comprobación de la estructura puede lograrse con los mismos métodos de degradación empleados en el estudio de los alquenos. La ozonólisis de dienos da aldehídos y cetonas, incluidos los de doble extremo que tienen dos grupos C=O por molécula. Por ejemplo: