Función del disolvente
(Ir a página principal http://organica1.org/ )
6.3 Solubilidad: solutos no
iónicos
6.4 Solubilidad: solutos
iónicos. Disolventes próticos y apróticos.
Pares iónicos
6.5 Reacción SN1:
función del disolvente. Enlaces ión-dipolo
6.6 Reaccción SN2:
función del disolvente.
Disolventes próticos y
apróticos.
6.7 Reacción SN2:
catálisis por transferencia de fase
6.8 SN2 contra SN1:
efecto del disolvente
6.9 Solvólisis. Ayuda
nucleofílica del disolvente
Enlace secundario
6.1
Función del disolvente
Como dijimos al comenzar el capítulo 5, la
mayor parte de la química orgánica es
heterolítica, y este tipo de reacciones se realiza en solución. Si queremos
comprender las reacciones orgánicas, entonces debemos aprender algo sobre la
función de un componente del sistema de reacción que raramente aparece en las
ecuaciones, pero que casi siempre está
presente: el disolvente.
El papel del disolvente no es pequeño. El
estudio de reacciones heterolíticas en ausencia de un disolvente en fase
gaseosa nos ha dado un patrón que nos muestra lo considerables que pueden ser
los efectos del disolvente. La presencia de un disolvente puede acelerar o incluso retardar una reacción en un factor
de 1020; un cambio de un disolvente a otro puede modificar la
velocidad de la reacción en casi un millón de veces. Los efectos del disolvente
pueden ser más poderosos que los otros factores; mucho más que los efectos
polares o estéricos, e incluso algo más
poderosos que los efectos de sinforia, que estudiaremos en el capítulo 20. El
disolvente la selección de un disolvente en particular puede ser el factor más
importante para determinar la rapidez
de una reacción e incluso si se realiza o no; puede determinar cuál de los distintos caminos alternativos
seguirá realmente una reacción.
Evidentemente, un disolvente no es
simplemente un lugar una especie de gimnasio donde las moléculas del soluto
pueden brincar y chocar ocasionalmente. El disolvente está íntimamente implicado en toda reacción que se
realiza en él, por lo que para nosotros
es importante comprender cuánto está implicado y de qué modo.
Hemos aprendido lo suficiente para darnos
cuenta de que las moléculas y iones del soluto no existe en solución como partículas
desnudas; están solvatadas (Sec.
1.21). Hay muchas moléculas de disolvente unidas por enlaces a cada
partícula disuelta y es la formación de
dichos enlaces la que proporciona la energía necesaria para que se rompan los
enlaces que mantienen unidas las partículas del soluto.
La ciencia de la química orgánica se basa en
una simple premisa: que la estructura molecular determina el comportamiento
químico. En solución, todos los participantes de una reacción química están
solvatados, tanto los reactivos como los productos e incluso el estado de transición. Nuestro estudio de
la reactividad química se basa en las diferencias energéticas entre los
reactivos y los estados de transición; es decir, estimamos las estabilidades
relativas de dichas especies. Para lograr esto, examinamos mental y, con el uso de modelos, físicamente
las estructuras implicadas; en este examen se deben incluir las aglomeraciones de disolvente que ayudan
a formar dichas estructuras y a determinar sus estabilidades.
En este capítulo, utilizando como ejemplos
las ya estudiadas reacciones de sustitución nucleofílica alifática, veremos
cómo se afecta la reactividad con el disolvente y con ella, el curso de la
reacción. El disolvente añade una nueva dimensión a nuestro estudio de la química
orgánica: si bien es cierto que complica las cosas, también es cierto que
resulta enriquecedor. Nos ofrece el modo más práctico para controlar lo que sucede en
una reacción química. El efecto que ejerce un disolvente es un tipo de efecto
del medio ambiental, y en ese sentido es el comienzo de una pista que conduce
hasta la reacción orgánica fundamental, la acción de una enzima; esta acción vital (literalmente) sólo es posible porque el sustrato se disuelve en la
enzima, quedando sujeto a ella por los mismos tipos de fuerzas que utiliza un
disolvente.
Empecemos pues el estudio de la función del
disolvente aprendiendo más sobre los
tipos de enlaces que se forman y rompen cuando ocurre la disolución.
6.2
Enlace secundario
En nuestro análisis del capítulo 1 sobre los
puntos de fusión y ebullición, y la solubilidad, se vieron brevemente los tipos
de fuerzas que actúan entre moléculas,
entre iones y entre moléculas y iones, que, como ya dijimos, son fuerzas
electrostáticas la atracción entre lo positivo y lo negativo. Dichas fuerzas de
atracción enlaces son las siguientes:
(a) Enlaces ión-ión: la atracción entre las cargas opuestas de un catión y un anión.
![]()
(b) Enlaces dipolo-dipolo: la atracción entre el extremo positivo de una molécula polar y el
negativo de otra, también polar.

De los enlaces dipolo-dipolo el más fuerte es
el puente de hidrógeno en el cual un
átomo de hidrógeno actúa como puente entre dos átomos electronegativos (F, O,
N), sujetando a uno donador del
hidrógeno-puente con un enlace covalente,
y al otro aceptor del hidrógeno-puente, por atracción puramente
electrostática. Por ejemplo:
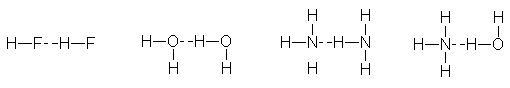
La fuerza de un puente de hidrógeno
determinado depnde de la naturaleza del donador y del aceptor del
hidrógeno-puente. Cuanto más ácido es el
donador, más fuerte es el puente de hidrógeno. La fuerza de dicho puente
depende de lo positivo que sea el hidrógeno (Sec. 1.19), y la acidez depende de
la capacidad que tenga la base conjugada para acomodar el par de eelctrones que
deja atrás el protón saliente. Ambas propiedades aumentan en el mismo factor:
la extracción de electrones en el grupo que está unido al hidrógeno. Cuanto más básico es el aceptor , más fuerte
es el puente de hidrógeno. La fuerza de un punete de hidrógeno depende de
lo negativo que sea el átomo aceptor; es decir, de lo disponibles que están sus electrones, esa disponibilidad es
lo que hace básica a una molécula.
(c) Fuerzas de Van der Waals: es la atracción entre los extremos con cargas opuestas de dipolos
momentáneos e inducidos en moléculas vecinas. Estas fuerzas actúan entre todas
las moléculas, incluidas las no polares.
(d) Enlaces ión-dipolo:
es la atracción de un ión positivo por el extremo negativo de las moléculas de
un disolvente polar, y de un ión negativo por el extremo positivo.

Excepto para los enlaces totalmente iónicos
de un cristal iónico, estas fuerzas de atracción se suelen denominar enlaces secundarios y actúan entre diferentes moléculas y iones, en
contraste con los enlaces covalentes que actúan dentro de una molécula o ión, manteniendo juntos sus átomos.
Individualmente, los enlaces secundarios son relativamente débiles, pero al
actuar al mismo tiempo como un equipo un conjunto de dichos enlaces es muy
fuerte y su formación, como veremos, puede proporcionar la energía suficiente
para romper un enlace covalente.
Si esto resulta sorprendente, hay que
recordar que para lograr que el cloruro de sodio hierva, debemos calentarlo a
1413ºC; a temperatura ambiente, podemos disolverlo en pocos minutos agitándolo
en un reciepiente con agua. Sin embargo, las fuerzas interiónicas que se vencen
en ambos procesos son exactamente iguales.
En este capítulo estaremos interesados
principalmente en los enlaces secundarios implicados en la acción del
disolvente: disolución de solutos, la influencia sobre su reactividad, e
incluso, de forma muy directa, su reacción con ellos; los enlaces secundarios
están involucrados en muchos más efectos, además de los del disolvente. Estas
mismas fuerzas, al actuar entre las moléculas alargadas y filiformas del
algodón, lana, seda y nailon proporcionan la fuerza necesaria para la formación
de las fibras (Sec. 36.8). Incluso las fuerzas de Van der Waals, que son las
más débiles y actúan entre las cadenas no polares de los fosfolípidos, son el cemento
de las paredes de las células vivas (Sec. 37.8).
Los enlaces secundarios existen tanto entre
moléculas diferentes, como entre distintitas partes de la misma molécula, y es
así como tienen una función clave en la determinación de las formas de moléculas grandes, como
proteínas y ácidos nucleicos, formas que, a su vez, determinan sus propiedades
biologicas; por ejemplo, el tamaño de las <<cavidades>> en la molécula de hemoglobina es suficientemente grande
como para contener los grupos hemo con
sus átomos de hierro acarreadores de oxígeno (Sec. 40.15); la forma espiral de
las moléculas de a-queratina y colágeno confiere resistencia a la lana y al
pelo, y fortalezca a los tendones y a la piel (Sec. 40.1). Los enlaces
secundarios son los responsables de que la espiral del ADN sea doble, lo que permite la autoduplicación
de moléculas, que es la base de la herencia (Sec. 41.8).
Por tanto, nuestro estudio en este capítulo
tendrá dos propósitos: entender mejor cuál es el papel del disolvente y, al
mismo tiempo, comprender mejor la naturaleza de los enlaces secundarios
6.3
Solubilidad: solutos no iónicos
Como se dijo anteriormente (Sec. 1.21), las
características de la solubilidad de los solutos no iónicos dependen principalmente de su polaridad y en particular
de su capacidad para formar puentes de hidrógeno. Nuestra regla empírica dice
que <<una sustancia disuelve a otra semejante>>.
Consideremos los tipos de compuestos con los
que nos hemos encontrado, comenzando por los hidrocarburos y los halogenuros de
alquilo, que son no polares o débilmente polares y se disuelven en disolventes
de polaridad semejante: en hidrocarburos como ligroína o benceno; en
halogenuros de alquilo como cloroformo o tetracloruro de carbono; en dietil
éter. Las fuerzas que unen entre sí a las moléculas del soluto y a las del
disolvente se reemplazan rápidamente por otras similares que mantienen unidas
las moléculas del soluto con las del disolvente. Los hidrocarburos y los
halogenuros de alquilo no se disuelven en agua, cuyas moléculas son muy polares
y se unen unas a otras fuertemente por puentes de hidrógeno.
Volvamos ahora a los alcoholes.
Estructuralmente, un alcohol es una mezcla de un alcano y agua: tiene un grupo
alquilo, semejante a un alcano, y un grupo hidroxilo, semejante al agua.
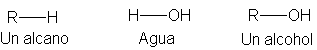
El grupo hidroxilo es muy polar y, lo que es
más importante, contiene un hidrógeno enlazado a un elemento altamente
electronegativo: el oxígeno. Mediante el grupo hidroxilo las moléculas de un
alcohol son capaces de formar puentes de hidrógeno: puentes entre sí mismas que
dan a los alcoholes sus puntos de ebullición anormalmente elevados (Sec. 1.20 y
17.5); puentes de hidrógeno con otras moléculas, que tienden a hacer los alcoholes más solubles en otros compuestos
hidroxilos, como el agua. Los alcoholes pequeños, como el metanol (CH3OH)
son completamente solubles en agua (Sec. 1.21). Los puentes de hidrógeno que
existen entre moléculas de agua y metanol pueden reemplazar fácilmente a los
muy similares puentes de hidrógeno formados entre diferentes moléculas de
metanol y diferentes moléculas de agua.
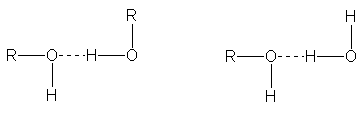
Debido a la condición muy especial del agua
como disolvente especialmente en sistemas biológicos se utilizan las palabras hidrófilo (que ama al agua) e hidrófobo (que odia al agua) para designar
la solubilidad e insolubilidad en agua. A menudo se emplean el término lipófilo (que ama las grasas), en lugar
de hidrófobo; esto destaca no tanto la insolubilidad en agua, sino más bien la
solubilidad en disolventes no polares. Así, el metanol es hidrófilo y los
alcanos y halogenuros de alquilo son lipófilos (o hidrófobos).
Puesto que es más fácil trabajar con un
término correspondiente a una cualidad positiva que con uno para una cualidad
negativa, en este libro usaremos, generalmente, la palabra lipófilo, con la que queremos indicar simplemente el hecho de la solubilidad en disolventes
no polares. Bien puede ser, como sostienen muchos, que esa solubilidad se deba
más al rechazo por el agua que a la aceptación positiva por un disolvente no
polar.
Consideremos ahora el efecto del grupo
alquilo en la solubilidad de una serie de alcoholes en los que el grupo
hidroxilo es hidrófilo, y el grupo alquilo, lipófilo. La tabla tabla 6.1 da la
solubilidad en agua de una serie de alcoholes. Para los miembros inferiores de
la serie, el grupo OH representa una parte grande de la molécula, de modo que
estos compuestos son miscibles con agua. Sin embargo, podemos ver que a medida
que aumenta el número de carbonos, la solubilidad disminuye regularmente: una
cadena larga con un OH en un extremo es principalmente un hidrocarburo, y su
solubilidad lo indica (véase Fig. 6.2).
Ahora bien, si una molécula es grande
suficiente si un alcohol, por ejemplo, tiene una cadena de 16 a 20 carbonos o
más sus partes hidrófilas y lipófilas exhiben sus propiedades de solubilidad
individuales. Las partes hidrófilas y lipófilas exhiben sus propiedades de
solubilidad individuales. Las partes hidrófilas se disuelven en agua, y las
lipófilas, en un disolvente no polar
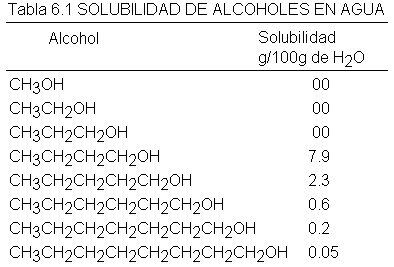
o, si
no hay ninguno presente, se agrupan de hecho, se disuelven mutuamente. Este
comportamiento de solubilidad dual confiere a los jabones y detergentes su
poder limpiador (Secs. 37.3 y 37.5) y controla la alineación de moléculas en
las membranas celulares (Sec. 37.8); una molécula de una proteína globular
digamos, una enzima se enrolla para exponer sus partes hidrófilas al agua
circundante y para esconcer sus fragmentos lipófilos; al hacer esto, adquiere
una forma muy particular, necesaria para sus propiedades biológicas
características (Sec. 40.11).
6.4
Solubilidad: solutos iónicos. Disolventes próticos y apróticos. Pares iónicos
Volvamos ahora a la disolución de compuestos
iónicos.
Las fuerzas que mantienen unido el retículo
iónico son poderosas, y para vencerlas se necesita una gran cantidad de
energía, que es suministrada por la formación de muchos enlaces ión-dipolo
entre los iones y el disolvente. Alrededor de cada ión se amontonan muchas
moléculas de disolvente, con su extremo positivo vuelto hacia un ión negativo,
y su extremo negativo, hacia un ión positivo (Fig. 6.1, Sec. 6.2).
Para disolver sustancias iónicas, un
disolvente debe ser altamente polar. Además, como hemos visto, debe tener una
elevada constante dieléctrica elevada,
es decir, debe ser un buen aislante para que disminuya la atracción entre los
iones con cragas opuestas una vez solvatados.
El agua debe su superioridad como disolvente
de sustancias iónicas sólo parcialmente
a su polaridad y a su elevada constante dieléctrica. Hay otros líquidos con
momentos dipolares considerables y altas constantes dieléctricas, y sin embargo
son disolventes muy pobres para compuestos iónicos. Lo que se necesita es poder de solvatación: la capacidad para
formar enlaces fuertes con los ioness disueltos. El poder de solvatación no
depende simplemente de un elevado momento dipolar, sino que tiene que ver con
la naturaleza de los enlaces ión-dipolo formados. Para ver lo que esto
significa, debemos observar más detenidamente la estructura del disolvente.
Comencemos con el agua.
Hemos establecido que los cationes son
atraídos al polo negativo de un disolvente polar.
En el agua, el polo negativo está en el
oxígeno, que es altamente electronegativo y, lo que es más importante, tiene pares de electrones no compartidos.
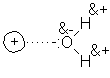
Además, con sólo dos minúsculos hidrógenos
unidos, el oxígeno está bien expuesto:
varios átomos de oxígeno de otras tantas moléculas de agua pueden agruparse en
torno al catión sin amontonarse.
Los aniones, dijimos que son atraídos al polo
positivo de una molécula polar. En el agua, los polos positivos se encuentran
claramente sobre los hidrógenos. Los enlaces ión dipolo que sujetan los aniones
al agua son puentes de hidrógeno.
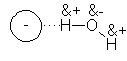
Estos enlaces de hidrógeno permiten una
solavtación particularmente intensa de los aniones: no sólo hay una carga
positiva grande concentrada sobre un átomo muy pequeño, hidrógeno, sino que
este hidrógeno sobresale de la molécula, estando muy bien expuesto. El anión
puede estar sujeto por varios puentes de hidrógeno a varias moléculas de agua
sin amontonarse.
Así, el agua debe una parte importante de su
notable poder de solvatación a su grupo OH: éste solvatada fuertemente a los cationes mediante los
pares de electrones no compartidos del oxígeno, y a los aniones, por puentes de
hidrógeno.
El metanol se parece al agua debido a su
grupo OH. No es de sorprender que también pueda disolver compuestos iónicos. (Sin embargo, es
inferior al agua: es menos polar, y el grupo CH3 es más grande y
ocasiona mayor aglomeración que el segundo H del agua).
Los disolventes como el agua y el metanol se
denominan disolventes próticos:
contienen hidrógeno unido a oxígeno o nitrógeno, de modo que son lo
suficientemente ácidos como para formar puentes de hidrógeno. Otros disolventes
próticos solvatan los iones del mismo modo que el agua: los cationes, mediante pares no compartidos, los aniones por medio de
puentes de hidrógeno.
En los últimos años se ha observado el
desarrollo y uso creciente de disolventes
apróticos: disolventes polares, de constante dieléctrica moderadamente
elevada y que no contienen hidrógenos ácidos. Por ejemplo:
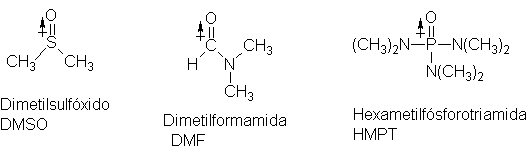
Estos disuelven compuestos iónicos, pero al
hacerlo su acción difiere de un modo muy importante de la de los disolventes
próticos: son incapaces de formar puentes
de hidrógeno con los aniones.
Estos disolventes apróticos son altamente
polares, con momentos dipolares varias veces mayores que el del agua. Como se
indica en las fórmulas, en cada uno de nuestros ejemplos el polo negativo se
halla sobre un átomo de oxígeno que sobresale
de la molécula (véase Figura 6.3). Los pares de electrones no
compartidos de estos átomos muy expuestos, cargados negativamente, pueden
solvatar los cationes muy fuertemente.
Por otra parte, el polo positivo se halla
sumergido dentro de la molécula. Debido a esta carga difusa y protegida, la
molécula sólo solvata los aniones muy débilmente. Por tanto, los disolventes apróticos disuelven compuestos iónicos principalmente
mediante la solvatación de cationes.
Como vimos en la sustitución nucleofílica,
gran parte de la química orgánica se ocupa de reacciones entre compuestos no
iónicos (generalmente orgánicos) y iónicos (tanto inorgánicos, como orgánicos),
por lo que es necesario elegir un disolvente en el que sean solubles ambos
reactivos. El agua disuelve muy bien sustancias íonicas, pero es un mal
disolvente para la mayoría de las orgánicas. Los disolventes no polares éter,
cloroformo, benceno disuelven bien compuestos orgánicos, pero son malos para
sales inorgánicas. Los alcoholes, en particular los más simple como metanol y
etanol, ofrecen una salida la tradicional a esta dificultad. Sus grupos
alquilo, lipófilos, les ayudan a
disolver reactivos orgánicos no iónicos; sus grupos hidroxilo les permiten
disolver reactivos iónicos. De esta manera, el metanol y el etanol
proporcionan, solos o mezclados con
agua, un medio en el cual se suelen realizar sustituciones nucleofílicas
alifáticas.
No obstante, el agua y los alcoholes son
disolventes próticos: solvatan aniones fuertemente mediante puentes de
hidrógeno, y resulta que generalmente los aniones son la parte importante de un
reactivo iónico. Así, aún cuando los disolventes próticos disuelven el
reactivo, poniéndolo en contacto con la molécula orgánica, estabilizan
simultáneamente los aniones, disminuyendo de forma radical su su reactividad;
se debilita su basicidad y, junto con esto su poder nucleofílico, una propiedad
relacionada (Sec. 5.9).
Es aquí donde actúan los disolventes
apróticos: mediante sus partes lipófilas, disuelven sustancias orgánicas;
también disuelven compuestos inorgánicos, pero logran esto, como acabamos de
ver, principalmente por solvatación de sus cationes. Los aniones quedan más o
menos libres y muy reactivos: son más básicos y más nucleofílicos.
En muchas reacciones se han logrado efectos
espectaculares al utilizar disolventes apróticos; reacciones que proceden
lentamente a temperaturas elevadas para dar rendimientos bajos en
disolventes próticos, pueden a menudo
ser muy rápidas muchas veces a temperatura ambiente, lográndose rendimientos
elevados si se realizan en un disolvente aprótico. El cambio a un disolvente
aprótico puede aumentar la velocidad de la reacción hasta un millón de veces.
Igual que los disolventes difieren en su
capacidad para solvatar iones, también los iones se diferencian en su tendencia
a ser solvatados. La carga
concentrada sobre un ión pequeño y duro conduce a enlaces ión-dipolo más
fuertes que la carga difusa de un ión más grande y <<blando>>. Así,
en un disolvente dado, se solvata más F- que CI-, y Li+
que Na+.
Hay una manera alternativa para visualizar la
estabilización de un ión por un disolvente.
De acuerdo con las leyes de la
electrostática, que estudiamos (Sec. 5.21),
la estabilidad de un sistema cargado aumenta por la dispersión de la carga. Consideremos
un anión solvatado, por ejemplo. Los extremos positivos de las moléculas del
disolvente están dirigidos hacia el anión, neutralizando su carga
parcialmente; al suceder esto, se
neutralizan parcialmente ellos mismos, lo que deja a las moléculas de
disolvente con una carga neta negativa, es decir, los extremos exteriores negativos ya no están exactamente
equilibrados por los extremos interiores positivos.
La carga negativa originalmente concentrada
sobre el anión, se halla ahora distribuida sobre la gran superficie externa del
conglomerado de molécula de disolvente. Esto significa una dispersión muy
considerable de la carga y, junto con ella, una estabilización enorme del
anión.
Por supuesto, los cationes se estabilizan de
igual forma por dispersión de su carga positiva sobre el disolvente.
Tal dispersión es más importante para la
estabilización de un ión pequeño, como F- o Li+, que para
un ión grande, como I- o Rb+, donde la carga ya se halla dispersa
sobre una superficie considerable.
La dispersión de carga por solvatación o
dentro del propio tiende a estabilizar cationes y aniones tanto orgánicos como
inorgánicos. Este concepto es clave para nuestra comprensión de una parte
importante de la química orgánica que involucra partículas intermediarias como
éstas, que hemos empezado a ver en nuestro estudio de los carbocationes en el
capítulo 5.
Hasta aquí sólo hemos estudiado la
interacción de un ión con el disolvente únicamente, pero hay otro componente de
la solución que debe ser considerado. Todo ión tiene un contraión, es decir, un ión
de carga opuesta, que está necesariamente presente. En soluciones acuosas
diluidas, un ión inorgánico está solvatado fuertemente y efectivamente aislado
de la carga de su contraión. Pero en un disolvente con menor poder de
solvatación o menor constante dieléctrica en metanol, por ejemplo, o en uno de
los apróticos descritos siente esta carga y es atraído por ella. Hay cierto
grado de enlace iónico y la pareja de iones de carga opuesta se denomina par iónico.
Las fuerzas de esta unión depende de la
naturaleza del disolvente. En disolventes en los que la solvatación es débil,
la unión iónica es fuerte: no hay moléculas de disolvente entre el par de
iones, por lo que hablamos de par iónico
compacto. En disolventes en los que la solvatación es más intensa, la unión
iónica es débil, pudiendo los iones estar separados por una o varias capas de
moléculas de disolvente, por lo que hablamos de un par iónico suelto.
Los pares iónicos tanto orgánicos como
inorgánicos son importantísimos en la química orgánica. Un ión en solución está
sometido a muchas fuerzas, y el efecto estabilizador de un contraión como
también del disolvente es algo que siempre debe ser tenido en cuenta.
Veamos ahora cómo interviene en las reacciones
químicas lo que acabamos de analizar.
6.5
Reacción SN1: función del disolvente. Enlaces ión-dipolo
Al estudiar cada una de las reacciones, SN2
y SN1, hemos explicado las diferencias en reactividad de varios
sustratos a partir de las diferencias en la cantidad de energía requerida: un
sustrato reacciona más rápido que otro principalmente a causa de una menor Eact.
En SN1, por ejemplo, la diferencia en la velocidad entre los
sustratos secundario y terciario corresponde a una diferencia en Eact
de unas 15 kcal.
Tratemos ahora una cuestión más básica una
que implica una cantidad mucho mayor de energía. ¿Cómo explicamos el hecho de
que haya sustitución incluso en el
caso de los sustratos más reactivos? Mediante cualquiera de los mecanismos, SN2
o SN1, se rompe un enlace entre un carbono y el grupo saliente el
enlace carbono-halógeno, por ejemplo, en un halogenuro de alquilo, y la ruptura
de un enlace requiere energía. ¿De dónde proviene esta energía?
Para una reacción SN2, la
respuesta es clara: la mayor parte de la energía necesaria para romper el
enlace al grupo saliente es proporcionada por la formación del enlace al
nucleófilo.
En el ataque mediante OH, digamos, se está
rompiendo el enlace carbono-halógeno y se está formando simultáneamente un
enlace carbono-oxígeno.
Pero, ¿qué podemos decir de la reacción SN1?
En este caso, el paso que determina la velocidad es una heterólisis
<<simple>> ruptura de enlace sin formación aparente de otro, para
equilibrar. En fase gaseosa, las energías de disociación de enlaces indican que
la heterólisis de un halogenuro de alquilo requiere una cantidad muy grande de
energía: 149 kcal/mol para el bromuro de t-butilo, y aún más para otros
sustratos. No obstante, la heterólisis en una reacción SN1 ocurre
con facilidad a temperaturas moderadas y con una Eact de sólo 20 a
30 kcal/mol. Esto deja una diferencia de unas 130 kcal o más, que hay que
proporcionar. ¿De dónde sale esta gran cantidad de energía?
Una vez más, la respuesta es dada por la
formación de enlaces: no de uno solo, como en la reacción SN2, sino
de muchos enlaces entre los iones
generador y el disolvente. No se
generan los iones como partículas desnudas en la casi vacía fase gaseosa, sino
en forma de iones solvatados.
Aglomerado alrededor de cada ión hay un grupo de moléculas polares de
disolventes, orientadas con sus extremos negativos hacia el carbocatión, y con
sus extremos positivos, hacia el anión (Fig. 6.4). Individualmente, estos
enlaces ión-dipolo son relativamente débiles, pero en conjunto proporcionan una
gran de energía.
Pero los iones son los productos de la heterólisis. Puesto que nos interesa la velocidad
de la hidrólisis, debemos considerar el estado de transición, no los productos,
y comparar su estabilidad con la del reactivo.
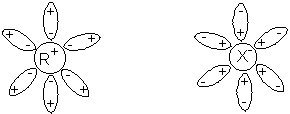
El reactivo tiene momento dipolar, y forma
enlaces dipolo-dipolo con moléculas de disolvente. (De hecho, se habría elegido
el disolvente, en parte, para este propósito, pues de otro modo el reactivo no
se disolvería.) El estado de transición tiene un enlace carbono halógeno
estirado con cargas positiva y negativa bien desarrolladas; tiene un momento

Dipolar mucho
mayorque el reactivo, por lo que forma enlaces dipolo-dipolo mucho más fuertes con el disolvente.
Este último estabiliza así al estado de transición más extensamente que al
reactivo, rebaja la Eact y acelera la reacción (Fig. 6.5). Al igual
que estabiliza los iones generados en una heterólisis, un disolvente polar
también estabiliza los iones incipientes en el estado de transición que origina
su formación. En una reacción SN1, la molécula del sustrato no se
descompone simplemente; se rompe por las moléculas del disolvente.
Lo que hemos venido tratando es la diferencia
entre heterólisis en ausencia y presencia de un disolvente. Es evidente que el
efecto de este último es enorme: rebaja la Eact en 130 kcal o más,
con lo que permite que ocurra la reacción.
Demos el próximo paso en nuestro análisis y
preguntémonos: ¿Cuáles son los disolventes que mejor promueven la heterólisis?;
esto es, ¿qué tipo de disolvente tiene el poder
ionizante más elevado? Para simplificar, tratemos la solvatación de los
productos, los iones, suponiendo que los mismos factores que los estabilizan
también estabilizan los iones incipientes en estado de transición. Partiendo de
esta base, el poder ionizante del disolvente depende entonces, en parte, de su polaridad: a igualdad de
otras cosas, cuanto más polar sea el disolvente, más fuertes serán los enlaces ión-dipolo.
Así, las reacciones SN1 de sustratos neutros son más rápidas en agua
que en etanol; van más rápidas en etanol al 20% (una mezcla 20:80 etanol:
agua), por ejemplo, que en etanol al 80%.
Pero ya vimos en la sección 6.4 que hay
muchas más cosas implicadas que la polaridad.
En general, los cationes se solvatan por
medio de pares de electrones no compartidos, mientras que los aniones se
solvatan esencialmente por puentes de hidrógeno. Ahora bien, aquí, los cationes
son carbocationes, y debido a la
dispersión de sus cargas forman enlaces ión-dipolo más débiles que los cationes
metálicos más pequeños. En consecuencia, en la ionización de estos sustratos
orgánicos, la solvatación del catión es relativamente débil, cualquiera que sea
el disolvente; es la solvatación del
anión lo que es especialmente importante. Para ello necesitamos disolventes
capaces de formar puentes de hidrógeno, es decir, disolventes próticos. Así, las reacciones SN1 se
realizan más rápidamente en agua, alcoholes y mezclas de agua y alcoholes que
en disolventes apróticos como DMF, DMSO y HMPT.
Podemos abarcar aún más, entre los
disolventes próticos, el poder ionizante es mayor para los que forman los
puentes de hidrógeno más fuertes, es
decir, los disolventes con los hidrógenos más ácidos. Por ejemplo, debido a la
gran atracción que ejercen los ´`atomos de flúor sobre los electrones, el
2,2,2-trifluoroetanol (CF3CH2OH) es más ácido que el
etanol; forma puentes de hidrógeno más fuertes con el grupo saliente y es mejor
disolvente para reacciones SN1. De la misma forma, los ácidos
fórmico (HCOOH) y trifluoroacético (CF3COOH) son excelentes
disolventes ionizantes.
Lo que hemos analizado en esta sección, por
tanto, es cómo el disolvente promueve la heterólisis rompiendo las moléculas de
sustrato. En la sección 6.9 veremos que el disolvente puede, a veces, hacer más
que tirar, también puede empujar.
6.6
Reaccción SN2: función del disolvente.
Disolventes
próticos y apróticos.
![]() Pasemos ahora a la reacción SN2, y veamos cómo la afecta el
disolvente. Consideremos el que es con mucha diferencia el más común de los
sistemas, aquél en el que el sustrato es una molécula neutra, y el nucleófilo,
un anión: la reacción de un halogenuro de alquilo con el ión hidróxido, por
ejemplo.
Pasemos ahora a la reacción SN2, y veamos cómo la afecta el
disolvente. Consideremos el que es con mucha diferencia el más común de los
sistemas, aquél en el que el sustrato es una molécula neutra, y el nucleófilo,
un anión: la reacción de un halogenuro de alquilo con el ión hidróxido, por
ejemplo.
Comencemos
del mismo modo que con la SN1, y veamos la diferencia entre una
reacción según se hace normalmente, en solución, y la realizada en fase gaseosa
es decir, sin nada de disolvente. Una vea
más se observa que el disolvente ejerce un efecto poderoso pero en sentido opuesto. Mientras el
disolvente acelera enormemente una reacción SN1, retarda la SN2, y en un
factor de hasta 1020.
¿Cómo
podemos explicar esta gran disminución en la velocidad? Como siempre que
tratamos un efecto que actúa sobre la velocidad de una reacción, debemos
comparar los reactivos con el estado de transición; esta vez, estudiaremos cómo
se ve afectado cada uno de ellos por el disolvente. Por definición, se
consideran dos reactivos en la etapa determinante de la velocidad de una
reacción SN2: en este caso, el halogenuro de alquilo y el ión
hidróxido.
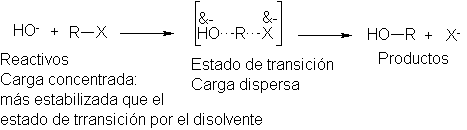
El
halogenuro de alquilo tiene un momento dipolar y establece un enlace
dipolo-dipolo débil con el disolvente. El ión hidróxido es portador de una
carga negativa neta, y forma enlaces ión-dipolo muy poderosos con el
disolvente. El estado de ransición también tiene una carga negativa neta, pero
aquí la carga está dividida entre el hidroxilo atacante y el halogenuro
saliente.El enlace del disolvente con esta carga dispersa es mucho más débil
que con la carga concentrada del pequeño
ión hidróxido. Así, el disolvente estabiliza los reactivos especialmente
el nucleófilo más que el estado de transición; eleva la Eact y
retarda la reacción (Fig.6.6).
La
solvatación del nucleófilo aniónico es el factor preponderantemente aquí.
Estabilizándolo en relación con el estado de transición, el disolvente desactiva el nucleófilo. La
desactivación del nucleófilo por el disolvente se ha medido molécula a
molécula. La reacción del bromuro de metilo y con iones hidróxido en fase
gaseosa se ha estudiado en diversos grados de hidratación, y se obtuvieron los
resultados siguientes:
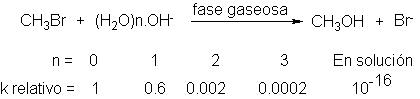
Partiendo
de un sistema exento de agua, observamos que, a medida que aumenta el número de
moléculas de agua por ión hidróxido, disminuye progresivamente la velocidad;
finalmente, en solución, la velocidad cae hasta una fracción muy pequeña de su
valor original.
No
obstante, la fuerza de la solvatación varía de un anión a otro, lo mismo que la
desactivación que ocasiona. Consideremos la reacción del bromuro de metilo con
varios iones halogenuro:
![]()
En
fase gaseosa, el orden de reactividad de los iones halogenuro es F- >
CL > Br- > I-, lo que refleja la fuerza del
enlace C-X que se está formando. En solución metanólica, en cambio, se invierte el orden de reactividad,
resultando I- > Br- > CI-
> F-.
La
explicación es evidente. La fuerza de la solvatación varía de un anión a otro,
y con ella, el gardo de desactivación. El fluoruro es el halogenuro más
pequeño, con la carga más concentrada; forma los enlaces ión-dipolo más fuertes
(Sec. 6.4) –puentes de hidrógeno en metanol, por lo que es el más desativado.
El yoduro es el más grande de estos halogenuros, con una carga dispersa; es el
menos fuertemente solvatado y, por tanto, el menos desactivado. En el metanol
no estamos comparando iones desnudos fluoruro y yoduro: estamos comparando un ión
fluoruro fuertemente solvatado con un yoduro cuya solvatación es débil. El
yoduro pertenece a la reacción más
rápida, no por su mayor reactividad intrínseca como alguna vez se creyó, sino
por estar menos solvatado. El disolvente es parte integral de la estructura de
una molécula disuelta; el ión fluoruro en metanol es un reactivo diferente del ión fluoruro en fase gaseosa o, para el
caso, de un ión fluoruro en DMF. Observamos dos órdenes de reactividad
diferentes para la reacción con bromuro de metilo, porque estamos en presencia
de dos conjuntos distintos de nucleófilos: no solvatados y solvatados.
Hasta
aquí, hemos tratado la diferencia entre una reacción SN2 en ausencia
y en presencia de un disolvente. Ahora bien, ¿Cuál es el efecto de cambiar de
un disolvente a otro?
En
general, entre disolventes similares, a mayor polaridad, mayor lentitud de una
reacción SN2; la estabilización por el disolvente más polar es más
fuerte para el nucleófilo aniónico que para el estado de transición, y aumenta
la Eact. (Una vez más, esto es lo opuesto a lo observado para una
reacción SN1.)Pero únicamente, estos efectos de la polaridad no son
muy grandes. En contraposición, los efectos de cambiar de un disolvente prótico
a uno aprótico son espectaculares. Las reacciones SN2 en disolventes
como dimetilsulfóxido (DMSO), dimetilformamida (DMF) o hexametilfósforotriamida
(HMPT), son hasta un millón de veces más
rápidas que en un alcohol o en una mezcla alcohol-agua. Nuevamente, es de
suma importancia la solvatación del anión: cuanto más fuertemente solvatado se
encuentre en relación con el estado de transición, más lenta será la reacción.
La solvatación más fuerte de aniones se hace por puentes de hidrógeno algo que
es posible en disolventes próticos,
pero no en los apróticos. Los disolventes apróticos disuelven reactivos iónicos
sobre todo por sus enlaces al catión: dejan al anión relativamente libre y muy
reactivo.
No
debemos olvidar que, al menos implícitamente, estamos considerando efectos
sobre el anión en relación con los
del estado de transición. Se justifica concentrar nuestra atención sobre el
anión, sencillamente por que la solvatación es más importante aquí y,
usualmente aunque no siempre, también
lo son las diferencias en la solvatación.
6.7 Reacción SN2: catálisis por transferencia de
fase
Al
cambiar de un disolvente prótico a uno aprótico, hemos dado, por tanto, un paso
en dirección hacia el medio <<ideal>> de reacción SN2;
la fase gaseosa, en la cual el anión se encuentra totalmente libre y es muy
reactivo. No obstante, incluso un disolvente aprótico solvata aniones, pues es
polar, de modo que establece enlaces ión-dipolo. Desde el punto de vista único
de la reactividad del nucleófilo, podríamos imaginar que un disolvente ideal
sería uno de polaridad muy baja, como un hidrocarburo o halogenuro orgánico:
benceno (C6H6), o cloruro de metilo (CH2CI2),
por ejemplo. Sin embargo, el propósito del disolvente es juntar los reactivos;
el sustrato orgánico se disolvería en tal disolvente, pero el reactivo iónico,
no. Este problema como el reactivo parece insoluble, pero ¿lo es?
Como
ejemplo, consideremos la reacción de un halogenuro de alquilo con cianuro de
sodio. El cianuro es fuertemente básico, anión nucleofílico, y desplaza
halogenuro para generar el cianuro de alquilo o nitrilo. (Veremos en Sec. 23.8 que éste es un paso importante en
la síntesis de ácidos carboxílicos.) La forma tradicional para llevar a cabo
esta reacción sería utilizar un disolvente prótico o aprótico que disuelva
ambos reactivos.

En
cambio, supongamos que tenemos una solución del halogenuro de alquilo en un
disolvente orgánico no polar y una solución de cianuro de sodio en agua, y
mezclamos ambas. Los disolventes son inmiscibles y se separan en dos capas dos fases. Podemos calentar esta mezcla
durante un tiempo muy largo, pero no sucederá nada. El sustrato permanece en la
capa orgánica y el nucleófilo queda en la fase acuosa, no pudiendo hacer lo que
deben, si han de reaccionar: no pueden
sufrir colisiones.
A
continuación, añadimos a esta mezcla una cantidad pequeña de una sal de amonio cuaternaria, un compuesto,
en el que se han reemplazado los hidrógenos del ión amonio por grupos alquilo
metilo o, mejor aún, grupos n-butilo. Para simplificar, denominaremos a este
catión cuat (Q+). Por
razones que se aclararán luego, el anión de esta sal podría ser bisulfato, HSO4-.
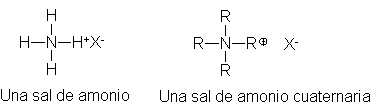
Y
ahora sucede algo notable: en presencia de una cantidad catalítica de esta sal
cuat, el halogenuro de alquilo y el cianuro reaccionan en apariencia aún
separados, cada uno en su propia fase-velozmente y en condiciones suavez, con
un elevado rendimiento en nitrilo.
Este
es un ejemplo de lo que Charles M. Starks (Continental Oil Company), uno de los
pioneros en este campo, ha denominado catálisis
por transferencia de fase. Pero, ¿cómo opera? Starks ha resumido el ciclo
catalítico como se muestra en la figura 6.7. Todo depende del hecho que los
grupos alquilo del ión cuat lo hacen lipófilo y, por tanto, capaz de entrar en
la fase orgánica. Sin embargo, no puede ir solo: para equipar su cara positiva
debe llevar consigo un anión. Este anión será ocasionalmente su contraión
original, o sea, bisulfato; este anión débilmente básico carece virtualmente de
poder nucleofílico, de modo que no hace nada.
Sin
embargo, la mayoría de los aniones en fase acuosa son iones cianuro (o
cualquier otro nucleófilo que se esté empleando), de modo que son los que
tienen mayor probabilidad de ser conducidos a la fase orgánica. Tenemos ahora
iones cianuro en un medio muy pocoprobable: un disolvente no polar. Allí, su
concentración puede ser muy baja, pero están virtualmente no solvatados y son
muy reactivos. La sustitución es rápida, formándose el nitrilo y liberándose un ión halogenuro.
Este ión halogenuro es conducido a la fase acuosa por el ión cual en su
migración de regreso.
Y
así continúa la reacción. El ión se mueve entre las dos fases transportando
aniones: a veces, el contraión original, otras, uno de los iones halogenuro
desplazados, y otras, el nucleófilo, un ión cianuro. Cuando sucede esto último,
puede haber reacción. La catálisis se debe así a la transferencia del nucleófilo de una fase a otra.
Hay
otro factor adicional involucrado en esto. Como hemos dicho, en la mayoría de
los disolventes, las sales existen en cierta medida, como pares iónicos (Sec. 6.4). Un ión siente la carga opuesta de su
contraión, y es atraído por ella. Cuanto menos polar es un disolvenet esto es,
cuanto más débil la solvatación, más fuerte es el apareamiento de iones: un
tipo de enlace es reemplazado por otro. También esta atracción electrostática
tiende a estabilizar un anión, y hacer esto, desactiva el anión como nucleófilo
y como base. Podríamos pensar que, al pasar a un disolvente no polar, estamos
simplemente intercambiando un tipo de desactivación por otro.
Sin
embargo, encontramos aquí otra ventaja del ión cuat como catalizador de
transferencia de fase: los grupos alquilo que lo hacen lipófilo son voluminosos
y protegen la carga positiva del nitrógeno, aislándola del anión (véase Fig.
6.8). El anión es atraído con menor intensidad hacia esta carga sumergida en el
ión cuat que hacia una carga concentrada sobre un catión metálico. Este par
iónico es muy suelto, y el anión es comparativamente libre y muy reactivo.
El
poder de la catálisis por transferencia de fase está entonces en el hecho de
que minimiza las dos principales fuerzas desactivadoras que actúan sobre un
anión: la solvatación y el apareamiento de iones. Existen muchas variantes del
método. No es necesaria una fase acuosa: puede transferirse el ión cianuro a la
fase orgánica directamente desde el cianuro de sodio sólidos. Tampoco se
necesita la presencia de un disolvente orgánico: el propio sustrato, si se
trata de un líquido, puede actuar de disolvente. El agente de transferencia de
fase no tiene que ser necesariamente iónico, sino que puede ser una molécula
neutra. Entre los catalziadores neutros, los más importantes son, como veremos,
los éteres corona, y con su estudio
entramos en el área fascinante de las relaciones anfitrión-huésped (Sec. 19.10).
En
sus diversas formas, la catálisis por transferencia de fase ha iniciado una
revolución en la técnica para llevar a cabo reacciones orgánicas, tanto en
laboratorio como en la industria: no sólo en la sustitución nucleofílica, sino
en reacciones de todo tipo eliminación, adición, oxidación, reducción. Esto le
ha dado al químico orgánico un arma nueva y poderosa para ayudarle a alcanzar
una de las metas principales, el control de
la reacción orgánica.
6.8
SN2 contra SN1: efecto del disolvente
Como
ya vimos, la sustitución nucleofílica puede efectuarse a través de dos
mecanismos diferentes, SN2 y SN1; en este capítulo hemos
analizado el efecto del disolvente sobre cada uno de ellos. De hecho, en estos
efectos del disolvente observamos otra evidencia que prueba la existencia de dos mecanismos: es una
diferencia más entre los dos tipos de reacción, que, por su magnitud, es la más
notable de todas. Tenemos dos reacciones que, en fase gaseosa, difieren en
velocidad en un factor de enormes proporciones: una reacción es ilimitadamente lenta,
y otra, enormemente rápida. No obstante, el disolvente acelera una de ellas y
retarda la otra de manera tal que, al tratar con la química ordinaria de
soluciones, debemos ocuparnos de la competencia entre ambas.
En
la sección 5.24 analizamos esta competencia entre SN2
y SN1. Para un sustrato dado y en determinadas condiciones, nos
preguntamos: ¿Cuál será el camino que seguirá una reacción? Y ¿qué podemos
hacer, si es posible hacer algo, para conducir la reacción en una u otra
dirección?
El
problema se reduce a lo siguiente, el mecanismo que se siga depende de cuál de
las dos reacciones ocurra más rápido en el sustrato: el ataque del nucleófilo o
bien la heterólisis, para formar un carbocatión.
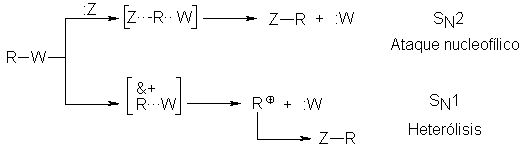
Ya
hemos analizado el sustrato y el
nucleófilo para ver qué factores determinan las velocidades relativas.
Consideremos ahora el tercer componente del sistema de la reacción, el disolvente.
Para
predecir el efecto del disolvente sobre la reacción mediante uno u otro
mecanismo, debemos comparar los reactivos con el estado de transición para el
sistema en particular involucrado. Tomemos el tipo más común de sustitución
nucleofílica: el ataque de un nucleófilo aniónico sobre un sustrato neutro.
Este sistema se examinó en detalle en las secciones 6.5 a 6.7, y vimos que los
efectos del disolvente son muy distintos para las reacciones por los dos
mecanismos. Una reacción SN1 es favorecida por disolventes de
elevado poder ionizante, es decir, por disolventes polares, próticos, que
ayudan a que salga de la molécula el grupo saliente. La reacción SN2
es favorecida por disolventes que estabilizan (y desactivan) lo menos posible al nucleófilo aniónico:
disolventes apróticos o de polaridad baja, como en la catálisis por
transferencia de fase.
No
es accidental que, para ilustrar el efecto de la estructura sobre la
reactividad en las secciones 5.15 y 5.22, elijamos para SN2 una
reacción realizada en el disolvente aprótico DMF, y para SN1, una
reacción en el disolvente CF3COOH, poalr y fuertemente enlazante por
puentes de hidrógeno (y débilmente nucleófilo).
(En
el efecto del disolvente observamos en parte un factor ya comentado: la
naturaleza del nucleófilo. En un disolvente aprótico o en condiciones de
transferencia de fase, estamos proporcionando un nucleófilo más poderoso, lo
que favorece, por supuesto, a SN2.)
De
todos los componentes de este sistema reaccionante, como ya dijimos, el
disolvente es el que ofrece el campo más amplio para el control de la reacción.
En la elección del sustrato y del nucleófilo estamos limitados por nuestro
deseo de obtener un producto en particular, pero en la elección del entorno
para efectuar la reacción, se nos abre un campo de posibilidades que está
ampliando rápidamente: desde disolventes fuertemente ionizantes y débilmente
nucleofílicos, en un extremo, hasta los apróticos o de transferencia de fase,
en el otro.
En
esta sección se explicó el efecto del disolvente, en la competencia entre
reacciones que ocurren por los mecanimos SN2 y SN1. Continuemos con
el tema de la competencia, ya no entre dos mecanismos nítidamente separados,
sino entre los factores que, de hecho, determinan lo que ha de suceder: poder
nucleofílico, impedimento estérico y dispersión de carga. Encontraremos que el
disolvente tiene un papel muy importante (de hecho, el principal).
6.9
Solvólisis. Ayuda nucleofílica del disolvente
Hemos
dicho (Sec. 5.9) que la sustitución nucleofílica alifática es el área de la
química orgánica más ampliamente estudiada y la más intensamente polémica. El
aspecto particular en que se centra la mayor parte del estudio y de la polémica
es el caso especial en el cual el nucleófilo es el disolvente: la solvólisis.
No
se agrega ningún nucleófilo fuerte, de modo que, para muchos sustratos, la
solvólisis cae en la categoría que hemos denominado SN1; es decir,
la reacción procede mediante dos o más pasos, con formación intermediaria de un
catión orgánico. Este catión es el que se encuentra en el centro del problema:
su naturaleza, cómo se forma y cómo reacciona. Al estudiar la solvólisis,
estamos explicando todas las reacciones SN1 y, en muchos aspectos,
todas las reacciones que implican carbocationes intermediarios.
Quizá
la pregunta más importante a contestar sea la siguiente: ¿qué función tiene
exactamente el disolvente? ¿Se aglomera en torno al carbocatión y al anión y al
estado de transición que conduce a su formación ayudando así a la heterólisis
mediante la formación de enlaces ión-dipolo, o actúa una única molécula de
disolvente como nucleófiilo y ayuda a empujar al grupo saliente para que salga de
la molécula? (Véase Fig. 6.9.)
Los
dos extremos citados corresponden a nuestras descripciones de los mecanismos SN2
y SN1. Por tanto, ¿no podríamos resolver nuestro problema
sencillamente mediante un estudio cinético? ¿Depende o no la velocidad de la concentracióndel
nucleófilo? Aquí nos encontramos con el problema especial que presenta la solvólisis. El nucleófilo es el
disolvente, y la concentración de éste no varía
durante el transcurso de la reacción. Cualquiera que sea la función del
disolvente en el paso determinante de la velocidad, observamos una cinética de primer orden; la velocidad depende sólo
de la concentración del sustrato. La reacción podría ser completamente SN1.
Sin embargo, por lo que la cinética revela, también podría ser totalmente SN2;
la cinética podría ser de seudo-primer
orden, y la constante kobs de velocidad observada podría ser, efectivamente,
una constante de velocidad verdadera, multiplicada por la concentración del disolvente; esto es, kobs=
k[:S].
![]()
En
consecuencia, no podemos decir si la velocidad depende o no de la concentración
del nucleófilo. Este hecho es, sin lugar a dudas, el origen de gran parte del
intéres por la solvólisis:la dificultad del problema es un desafio para el
químico orgánico. No obstante, esto no es todo. En comparación con aniones como
hidróxido, alcóxidos o cianuro, los disolventes son nucleófilos débiles. Pueden
competir otras fuerzas en contra de la debilidad de su acción, competencia que
puede observarse y medirse: la fuerza ejercida por un grupo que migra en una
transposición, por ejemplo, o por el grupo saliente que acaba de salir. Los
nucleófilos suaves permiten la formación de carbocationes, y el estudio de
estas partículas siempre es fascinante.
Parece
evidente que el disolvente puede dar ayuda
nucleofilica la solvólisis. La magnitud de esta ayuda depende de:
(a) el poder nucleofílico del disolvente
(b) lo imperiosa que sea la necesidad de ayuda, y
(c) lo accesible que sea estéricamente el carbono para la molécula que
auxilia.
El
agua, el metanol y el etanol, por ejemplo, son frecuentemente nucleofilicos
esto es, para ser disolventes; el ácido acético (CH3COOH) es más
débil, y el ácido fórmico (HCOOH), aún más débil. El ácido trifluoroacético (CF3COOH),
el alcohol trifluoroetílico (CF3CH2OH) y el alcohol hexafluoroisopropílico
(CF3CHOHCF3)son muy débiles: los átomos de flúor, muy
electronegativos, atraen fuertemente electrones del oxígeno, disminuyendo así
su basicidad y poder nucleofílico.
La
realidad de sustratos terciarios apenas depende del poder nucleofílico del
disolvente, y bastante de su poder ionizante (Sec. 6.5). La formación de
cationes terciarios es relativamente sencilla, y necesita poca ayuda nucleofílica; en todo caso, la aglomeración
no alienta esa ayuda. La reactividad de sustratos secundarios depende tanto del
poder nucleofílico del disolvente como ionizante. La formación de cationes
secundarios es más dificil, por ello necesita mucha ayuda nucleofílica. Con la
mayoría de los sustratos primarios, la reacción es probablemente SN2:
un solo paso, con el disolvente actuando como nucleófilo.
Concentrémonos
entonces en los sustratos alquílicos secundarios. ¿Qué se entiende por ayuda nucleofílica? En primer lugar, se
distingue del ataque del tipo SN2 en que no conduce a la formación
del producto, sino de un catión intermediario. Luego, se diferencia de la
solvatación general en que está involucrada una sola molécula de disolvente, y
no un grupo de ellas. La molécula de disolvente ataca al sustrato por la parte
de atrás y, actuando como nucleófilo, ayuda a sacar al grupo saliente por
delante. Se genera un carbocatión o, más bien, algo con mucho carácter de tal .
Por atrás, cuelga la molécula de disolvente y, por delante, el grupo saliente.
Cada uno puede estar ligado al carbono mediante el solapamiento de un lóbulo de
un orbital p del carbono el orbital p vacío del carbocatión clásico. La
geometría es similar a la del estado de transición SN2, pero éste es
un intermediario y corresponde a un
mínimo energético en un gráfico del avance de la reacción. (Compárese Fig. 6.10
con Fig. 6.9). Si el grupo saliente es un anión, y si el disolvente es de
polaridad moderada, la unión entre catión y anión puede ser principalmente
electrostática, y se habla de un par
iónico.
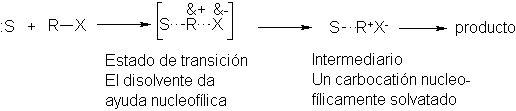
Este
intermediario catiónico este carbocatión nucleofílicamente solvatado reacciona
ahora. Se le abre amplia variedad de reacciones que, como veremos, pueden
sufrir los carbocationes. En la reacción que nos ocupa, se combina con una
molécula de disolvente con formación de un enlace total para generar el
producto. Si en el instante de esta reacción el grupo saliente todavía se
encuentra ligado a la cara frontal o aun acecha ahí, la reacción con el
disolvente ocurre por la parte de atrás. Por otra parte, si el catión ha durado
lo suficiente como para que el grupo saliente pudiera haber sido intercambiado
por una segunda molécula de disolvente generando así un intermediario
simétrico, la reacción es igualmente
probable por atrás que por delante. La solvólisis puede suceder con inversión
completa, o con inversión más cantidades variables de racemización.
El
elegante trabajo de Saul Winstein (Universidad de California, Los Angeles)
reveló el comportamiento detallado de
pares iónicos que son intermediarios en ciertos casos de solvólisis: pares firmes (o íntimos), cuyo catión es
suficientemente libre para girar y perder configuración, aunque suficientemente
sujeto para que la recombinación al compuesto enlazado covalentemente sea el
proceso favorecido; pares sueltos (o
separados por disolventes), cuyo catión es sucsceptible al ataque por
nucleófilos foráneos. El papel exacto que tienen los pares iónicos en la
sustitución nucleofílica ha sido objeto
de mucha investigación y, quizá, más debatido que el papel del disolvente;
ésta es, sin embargo, un área extensa de química complicada, por lo que no
podemos entrar en su estudio.
Se
ha dicho que existe un espectro continuo de mecanismos para la solvólisis, que
abarca desde la reacción SN1 clásica en un extremo, hasta la reacción SN2,
de un dolo paso, en el otro. En los gráficos de avance de la reacicón, el
mínimo energético para el carbocatión se hace cada vez menos profundo, a medida
que nos alejamos del extremo SN1; en el extremo SN2, ha
desaparecido el mínimo y queda un solo máximo.
Entre
los extremos del espectro hay mecanismos que implican grados variables de ayuda
nucleofílica de parte del disolvente. Paul Schleyer (Sec. 5.22), cuya
descripción de la ayuda nucleofílica corresponde esencialmente a la descrita
aquí, ha denominado SN2 (intermediario) a estos mecanismos; esto es,
son reacciones SN2 que involucran la formación de un intermediario.
Tal mecanismo tiene características de ambos mecanismos clásicos, el SN2
de una etapa, y el SN1. La terminología de Schleyer enfatiza el aspecto SN2:
el ataque nucleofílico proporciona una parte de la fuerza impulsora para la
reacción. Sin embargo, en este libro trataremos el mecanismo como si fuese una
modificación del SN1: se forma un intermediario catiónico uno que,
presumiblemente, es capaz de hacer todo lo que hace un carbocatión y la
dispersión de la carga positiva en
desarrollo proporciona gran parte de la fuerza impulsora de la reacción.
Consideraremos que la reacción sigue el mecanismo SN1 con ayuda
nucleofílica del disolvente, y al intermediario lo denominaremos carbocatión nucleofílicamente solvatado o, a
veces, carbocatión obstruido. Mientras nos entendemos, la terminología que
usamos no es importante. Los relevante está en apreciar la operación de los
mismos factores básicos reconocidos originalmente por Hughes s Ingold hace 50
años: ataque nucleofílico, con su susceptibilidad ante la obstrucción estérica,
y dispersión de la carga por medio de los sustituyentes y del disolvente. Lo
nuevo aquí es comprender lo importante que pueden llegar a ser ambos factores.
Debemos
mantener nuestro sentido de la perspectiva: estamos tratando el caso especial
de la solvólisis, y la mayoría de lo que decimos sólo tiene que ver con
sustratos de alquilos secundarios. Las diferencias en la estabilidad de las
distintas clases de carbocationes son suficientemente importantes como para
distinguir tres grupos diferentes: (a) para sustratos primarios, SN2
de un paso único: (b) para sustratos terciarios, SN1, con un
intermediario que se aproxima a nuestra idea de lo que es un carbocatión simple
(solvatado); (c) para sustratos secundarios, una reacción de dos pasos similar
a SN1 en la que existe un intercambio catiónico, pero formado con
ayuda nucleofílica y todavía obstruido con el nucleófilo (disolvente) y el
grupo saliente. La ayuda nucleofílica es un factor importante en la
determinación de reactividades relativas entre sustratos secundarios, y sus reactividades en diversos
disolventes pero también lo es el poder ionizante del disolvente. La ayuda
nucleofílica no es tan poderoso como la dispersión de carga que hace reaccionar
los sustratos terciarios sin ayuda nucleofílica más rápidamente que los
secundarios.
6.10
El medio: un mensaje
Lo
dicho en este capítulo sobre el control del emdio en el cual se efectúa la
sustitución nucleofílica es sólo el comienzo. Veremos que pueden realizarse
reacciones de muchas clases entre reactivos mantenidos en la esfera de
coordinación de los metales de transición (Secs. 20.5 a 20.8) o que residen
como huéspedes dentro de cavidades de
moléculas anfitrionas grandes, y
hechas a la medida (Secs. 19.10 y 39.10). Puede utilizarse el control del medio
de reacción para lograr reacciones nuevas o para acelerar las anteriores, y
para lograr un grado de selectividad
tanto en estereoquímica como en orientación y reactividad relativa que
anteriormente no era posible. No obstante, esto es sólo el principio.