Halogenuros de alquilo
Sustitución nucleofílica alifática
(Ir a página principal http://organica1.org/ )
5.1 Química homolítica y
heterolítica
5.2 Velocidad relativa de
reacciones competitivas
5.3 Estructura de
los halogenuros de alquilo.
5.4 Clasificación y
nomenclatura de los halogenuros de alquilo
5.5 Alcoholes: breve introducción
5.6 Propiedades físicas de los
halogenuros de alquilo
5.7 Preparación de halogenuros
de alquilo
5.8 Reacciones de los
halogenuros de alquilo.
Sustitución nucleofílica
alifática
5.9 Sustitución nucleofílica
alifática.
Nucleófilos y grupos
salientes
5.10 Velocidad de reacción:
efecto de la concentración. Cinética
5.11 Cinética de la sustitución
nucleofílica alifática.
Reacciones de
segundo y primer orden
5.12
Sustitución nucleofílica alifática: dualidad de mecanismos
5.13 Reacción SN2:
mecanismo y cinética
5.14 Reacción SN2:
estereoquímica, Inversión de la configuración
5.15 Reacción SN2:
reactividad. Impedimento estérico
5.16 Reacción SN1:
mecanismo y cinética.
Etapa
determinante de la velocidad
5.18 Estructura de
carbocationes
5.19 Reacción SN1:
estereoquímica
5.20 Estabilidades relativas
de carbocationes
5.21 Estabilización de
carbocationes. Acomodación de la carga. Efectos polares
5.22 Reacción SN1:
reactividad.
Facilidad
de formación de carbocationes
5.23
Transposición de carbocationes
5.25 Reacción de
alcoholes con halogenuros de hidrogeno.
Catálisis ácida
5.26
Análisis de halogenuros de alquilo
5.1 Química homolítica y
heterolítica
Como ciencia, la química debe su existencia,
por supuesto, al cambio químico: la conversión de una sustancia en otra. Moléculas
antiguas son transformadas en otras nuevas, lo cual significa que deben
romperse enlaces antiguos para generar otros nuevos; en su mayoría enlaces
covalentes, en lo que respecta a la química orgánica.
Hemos visto que la ruptura de un enlace covalente puede ocurrir de dos
maneras fundamentalmente diferentes (Sec. 1.14), dependiendo de lo que suceda
con los dos electrones que forman el par enlazante: en la homólisis, va un
electrón a cada fragmento; en la heterólisis, van ambos electrones al mismo fragmento.
Los sustantivos <<homólisis>> y heterólisis sólo se utilizan en su
sentido literal (Sec. 1.14), para indicar la ruptura de enlaces. No obstante,
los adjetivos <<homolítico>> y <<heterolítico>> a falta
de términos más adecuados se emplean en un sentido más general, para incluir
también el proceso de la construcción de enlaces, pudiéndose definir así dos
clases muy amplias de reacciones orgánicas.
Por consiguiente, las reacciones homolíticas son aquellas en las que se quitan o se
proporcionan los electrones del par enlazante individualmente, tanto si se rompen enlaces,
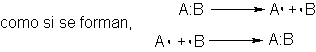
O si se rompen y forman simultáneamente,
![]()
Cada uno de los átomos que se separan lleva
uno de los electrones enlazantes, y
cada uno de los átomos que se juntan, proporciona uno de los electrones de
enlace.
Reaccciones
heterolíticas son aquellas en las cuales los electrones enlazantes se quitan o se
proporcionan en pares. Tanto si se
rompen enlaces.
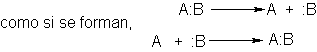
O si se rompen y forman simultáneamente,
![]()
Uno de los átomos que se separa lleva ambos
electrones de enlace, y uno de los átomos que se une proporciona el par.
.La química homolítica es, por tanto, la
química del electrón impar, mientras que la química heterolítica es la del par
de electrones. Mientras la primera se ocupa de partículas neutras, conocidas
como radicales libes, la segunda tiene que ver con cargas positivas y
negativas, es decir, con cationes y aniones. Las reacciones homolíticas suelen
producirse en la fase gaseosa, o en disolventes cuya función principal se reduce a proporcionar un medio
inerte que permita el movimiento libre de moléculas reaccionantes. Las
reacciones heterolíticas se realizan
generalmente en solución, y los disolventes tienen, como veremos, efectos muy
poderosos; la magnitud de su poder está empezando a conocerse ahora.
Hasta aquí, la reacción que más nos ha venido ocupando la sustitución
de radicales, ilustrada por la halogenación de los alcanos forma parte de la
química homolítica.
Comencemos ahora el estudio de la química heterolítica. La mayor parte
de la química orgánica es heterolítica, siendo ésta la que más trataremos en el
resto del libro. Tal como sucede con la halogenación, la reacción con la que
empezamos es una sustitución, pero de
un tipo muy diferente: es heterolítica y del tipo específico llamado sustitución nucleofílica alifática.
5.2 Velocidad relativa de reacciones competitivas
En el estudio de la sustitución nulceofílica ampliaremos enormemente
nuestra comprensión del principio que sustenta toda la química orgánica: que el
comportamiento químico depende de la estructura molecular. Antes de avanzar,
recordemos cómo, en general, abordamos la cuestión del comportamiento químico.
En un reactor hay una colección de moléculas, golpeando a ciegas en todas
las direcciones, chocando entre sí. Hay, en principio, varias opciones para
ellas: diversas reacciones que pueden llegar a sufrir. La que sucede realmente
o al menos predomina es aquélla que va
más velozmente. Por tanto, el comportamiento químico se reduce a una
cuestión de la velocidad relativa de
reacciones que compiten.
Como vimos en nuestro estudio de la halogenación de alcanos, las
velocidades relativas de las reacciones determinan:
(a) Qué sucede: un átomo de halógeno, por ejemplo, se une a un hidrógeno del
metano, extrayendo el hidrógeno; la alternativa, la unión al carbono con
expulsión de un átomo de hidrógeno, es muchísimo más lenta, y de hecho no
sucede (Sec. 2.21).
(b) Dónde sucede: un átomo de halógeno prefiere extraer un hidrógeno del etano,
antes que del metano; extrae un hidrógeno terciario con preferencia a uno
primario (Sección 3.23).
(c) Incluso, si sucede: un átomo de cloro extrae hidrógeno de un alcano,
mientras que uno de yodo, no, porque se combina más rápidamente con otro átomo
de yodo (Sec. 2.20).
Una reacción química es, en consecuencia, el resultado de una competencia: es una carrera ganada por
el corredor más veloz. Aprendimos, además, que el factor más importante que
determina la velocidad de una reacción es la energia de activación. Lo que
tiende a hacer nuestra colección de moléculas es, en general, lo que resulta más fácil para ellas:
siguen el curso que demanda menos energía, o sea, sufren la reacción con la
menor Eact.
Y, finalmente, para ayudarnos a comprender la Eact a
interpretarla y, a veces, incluso a predecirla disponemos de nuestra
importancia herramienta intelectual: el estado de transición. Cuanto más
estable sea el estado de transición en relación con el reactivo, menor será la
Eact y más rápida la reacción. El concepto del estado de transición
es nuestra unión mental entre la estructura molecular y el comportamiento
químico.
Lo expuesto anteriormente se basa en la premisa de que los productos
que obtengamos de una reacción química, y sus proporciones relativas, reflejan
la velocidad relativa a la que se forman inicialmente; es decir, una vez formado, un producto
particular se mantiene sin cambio hasta que se completa la reacción. Para la
mayoría de las reacciones que estudiamos, esta premisa es correcta; en las
condiciones utilizadas, la mayoría de las reacciones orgánicas son
esencialmente irreversibles, es decir, son
reacciones en un solo sentido.
Pero no siempre éste es el
caso. Algunas reacciones son reversibles, y existe un equilibrio entre los
diversos productos; lo que obtenemos
así, refleja el producto que resulta favorecido por el equilibrio, no el que
inicialmente se forma más rápido.
Veremos más adelante ejemplos de este tipo de comportamiento. En consecuencia,
para una reacción orgánica, la irreversibilidad no es algo que podamos dar
simplemente por supuesto; ha de ser comprobada experimentalmente, y sólo una
vez que haya sido comprobada estamos justificados en interpretar la composición
de los productos basándonos en la velocidad relativa.
En nuestro estudio de la sustitución nucleofílica tendremos mucho que
ver con la competencia entre posibles
vías de reacción: en este capítulo, la competencia entre diversos
mecanismos para la propia sustitución; en otros, la competencia entre
sustitución nucleofílica y una reacción de un tipo totalmente diferente, la
eliminación. Nos ocuparemos de la velocidad relativa a la que tienen lugar
estas reacciones competitivas y de las clases de estados de transición por los
que pasan. Lo que es más importante, estudiaremos los factores que determinan
la estabilidad de estos estados de transición, factores con los que operaremos
en el resto de nuestro estudio de la química orgánica.
5.3 Estructura de los
halogenuros de alquilo.
El grupo funcional
En esta introducción a la sustitución nucleofílica, trataremos
principalmente una familia de compuestos que ya nos resulta familiar, los halogenuros de alquilo. Los
halogenuros de alquilo tienen la fórmula general RX, donde R representa un
grupo alquilo o alquilo sustituido.
R-X
Un halogenuro de alquilo
La característica principal de la estructura del halogenuro de alquilo
es el átomo de halogenuro, X, y las reacciones características de un halogenuro
de alquilo son las que tienen lugar en el halógeno. El átomo o grupo que define la estructura de una familia particular de
compuestos orgánicos y, al mismo tiempo, determina sus propiedades se llama grupo funcional.
En los halogenuros de alquilo, el grupo funcional es el halógeno. No
debemos olvidar que un halogenuro de alquilo tiene un grupo alquilo unido a
este grupo funcional; en condiciones apropiadas, estas partes alquílicas
sufrirán las reacciones típicas de los alcanos. No obstante, las reacciones que
son características de la familia son
las que ocurren en el átomo de halógeno.
Por tanto, los diversos grupos funcionales son parte importante de la
química orgánica. Aprenderemos a asociar un conjunto particular de propiedades
con un grupo determinado dondequiera que lo hallemos. Cuando nos encontramos
con una molécula complicada que contiene varios grupos funcionales diferentes,
podemos esperar que las propiedades de esta molécula sean aproximadamente una
mezcla de las propiedades de los distintos grupos presentes. Un compuesto que
contiene X y OH es a un tiempo un halogenuro de alquilo y un alcohol;
dependiendo de las condiciones experimentales, puede sufrir reacciones
características de ambos tipos de compuestos. Las propiedades de un grupo
pueden verse modificadas, por supuesto, por la presencia de otro grupo, y para
nosotros es importante entender estas modificaciones; sin embargo, nuestro
punto de partida es la química de grupos funcionales individuales.
En este capítulo estudiaremos los halogenuros de alquilo de una forma sistemática. Delinearemos su química, para concentración luego en su reacción más importante: la sustitución nucleofílica.
5.4 Clasificación y
nomenclatura de los halogenuros de alquilo
Clasificamos un átomo de carbono como primario, secundario o terciario, según el número de otros átomos
de carbonos unidos a él (Sec. 3.11). Se clasifica un halogenuro de alquilo de
acuerdo con el tipo de carbono que sea portador del halógeno:
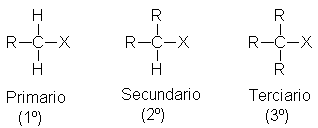
Como miembros de la misma familia, con el mismo funcional, los halogenuros
de alquilo de las diversas clases tienden a dar el mismo tipo de reacciones.
Sin embargo, difieren en la velocidad de reacción, y estas divergencias pueden
originar otras diferencias, más profundas.
Vimos (Sec. 3.8 y 3.10) que pueden darse dos tipos de nombres a los
halogenuros de alquilo: nombres comunes
(para los más sencillos), y nombres
IUPAC, con los que el compuesto sencillamente se denomina como un alcano
con un halógeno unido en forma de cadena lateral. Ejemplos:
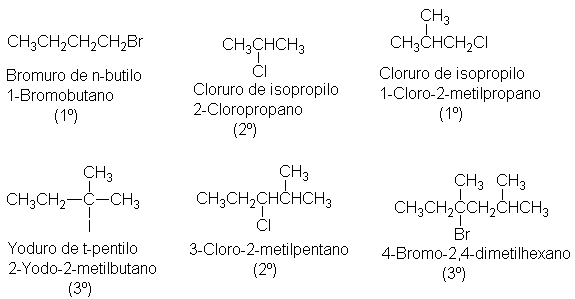
Ha de tenerse en cuenta que nombres similares no siempre significan
igual clasificación: cloruro de isopropilo, por ejemplo, es un cloruro
secundario, mientras que cloruro de isobutilo es primario.
5.5 Alcoholes: breve introducción
Nos podemos avanzar mucho en la química orgánica de la mayoría de los
grupos sin encontrarnos con otra familia de compuestos: los alcoholes. La química de los halogenuros de alquilo no es la
excepción. Para comenzar, los alcoholes son las sustancias a partir de las
cuales casi siempre se forman los
halogenuros de alquilo, y de hecho la mayoría de los compuestos que sufren sustitución nucleofílica. Ahora bien, una
vez que se obtienen halogenuros de alquilo y se someten a sustitución
nucleofílica, nuevamente nos encontramos con los alcoholes desempeñando gran
variedad de papeles: pueden ser los reactivos con los cuales reaccionan los
halogenuros de alquilo, los productos o los disolventes en los que tiene lugar
la reacción. (De hecho, veremos que la preparación de halogenuros de alquilo
involucra también sustitución nucleofílica, esta vez con alcoholes como
reactivos sometidos a sustitución.)
Más adelante estudiaremos sistemáticamente la química de los alcoholes
(Caps. 17 y 18), pero por ahora aprenderemos justo lo suficiente sobre estos
compuestos para poder trabajar con ellos.
Los alcoholes son compuestos de fórmula general R-OH, donde R es
cualquier grupo alquilo y OH es el grupo hidroxilo. Un alcohol simple puede
nombrarse dando el nombre del grupo alquilo que está unido al grupo hidroxilo
con la terminación ílico, precedido de la palabra alcohol. Los nombres IUPAC se derivan agregando una letra l final
al nombre del alcano correspondiente. (Esta, nomenclatura se trata
detalladamente en la Sec. 17.4, que puede consultarse si se considera
necesario.) Al igual que un halogenuro de alquilo, un alcohol se clasifica como
primario, secundario o terciario,
según el tipo de carbono que lleva el grupo OH.
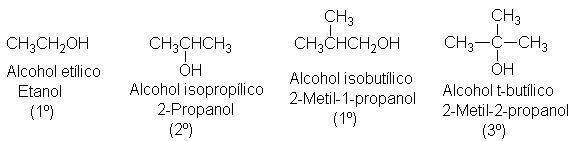
Entre las propiedades de los alcoholes, destaca un par de ellas que debemos
conocer: su acidez y su basicidad.
Estas propiedades residen en el grupo hidroxilo, OH, el grupo funcional de los
alcoholes. Este grupo es similar al grupo hidroxilo del agua, un compuesto que
ya nos resulta familiar. Los alcoholes, al igual que el agua, son ácidos y
bases débiles, casi tan ácidos y tan básicos como el agua.
Lo mismo que el agua, los alcoholes son lo suficientemente ácidos como
para reaccionar con metales activos, liberando hidrógeno gaseoso
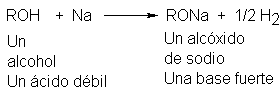
Los productos formados se llaman alcóxidos:
por ejemplo, etóxido de sodio o n-propóxido de potasio. Los alcóxidos son bases
fuertes, al igual que el hidróxido de sodio.
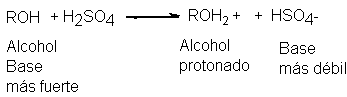
Al igual que el agua, también los alcoholes son lo suficientemente
básicos como para aceptar un protón de ácidos fuertes como el cloruro y el
sulfato de hidrógeno, permitiendo así la disociación completa de estas
sustancias, por ejemplo:
5.6 Propiedades físicas de los halogenuros de
alquilo
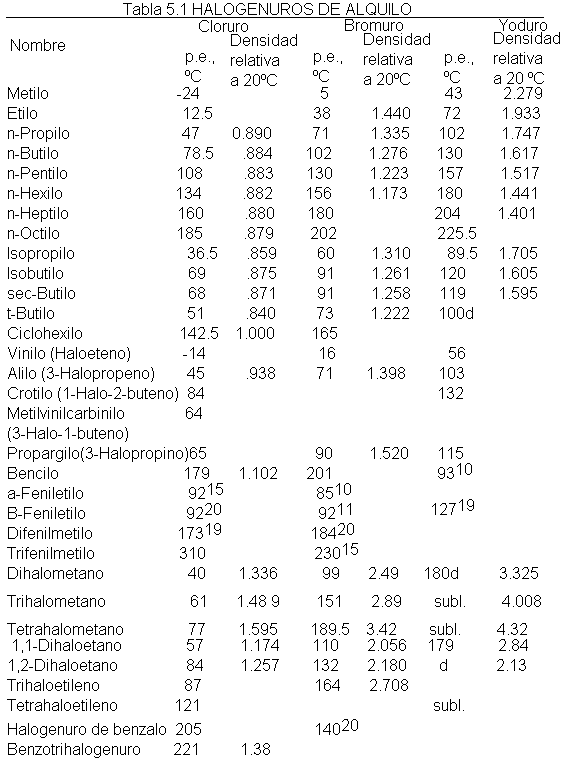
Debido a su mayor peso molecular, los haloalcanos tiene puntos de
ebullición considerablemente más altos que los alcanos de igual número de
carbonos (Tabla 5.1). Para un grupo alquilo dado, el punto de ebullición
aumenta con el incremento en el peso atómico del halógeno, de modo que un
fluoruro hierve a la temperatura más baja, y un yoduro, a la más elevada.
Para un halógeno determinado, el punto de ebullición aumenta al
aumentar el número de carbonos: al igual que en los alcanos, el aumento de
punto de ebullición es de unos 20-30 grados por cada carbono adicional, salvo
en el caso de los homólogos muy pequeños. Igual que antes, el punto de
ebullición disminuye con el aumento de las ramificaciones ya impliquen un
grupos alquilo o al propio halógeno.
A pesar de sus modestas polaridades, los halogenuros de alquilo son
insolubles en agua, tal vez porque no son capaces de establecer puentes de hidrógeno. Son solubles en los
disolventes orgánicos típicos, de baja polaridad, como benceno, éter,
clororformo o ligroína.
El yodo, el bromo y los policlorocompuestos son más densos que el
agua.
En consecuencia, los alcanos y halogenuros de alquilo tienen las
propiedades físicas que podemos esperar para compuestos de baja polaridad,
cuyas moléculas se mantienen juntas por fuerzas de Van der Waals o por
atracciones dipolares débiles. Tienen puntos de fusión y de ebullición
relativamente bajos, y son solubles en disolventes no polares e insolubles en
agua.
Hay otra consecuencia que resulta de su baja polaridad: mientras los
alcanos y halogenuros de alquilo son buenos disolventes para otras sustancias
de baja polaridad entre ellos mismos, por ejemplo, son incapaces, en cambio, de
solvatar apreciablemente iones simples, de modo que no pueden disolver sales
inorgánicas
5.7 Preparación de
halogenuros de alquilo
En el laboratorio, los halogenuros de alquilo se suelen preparar por los métodos expuestos a continuación.
PREPARACION DE HALOGENUROS DE ALQUILO
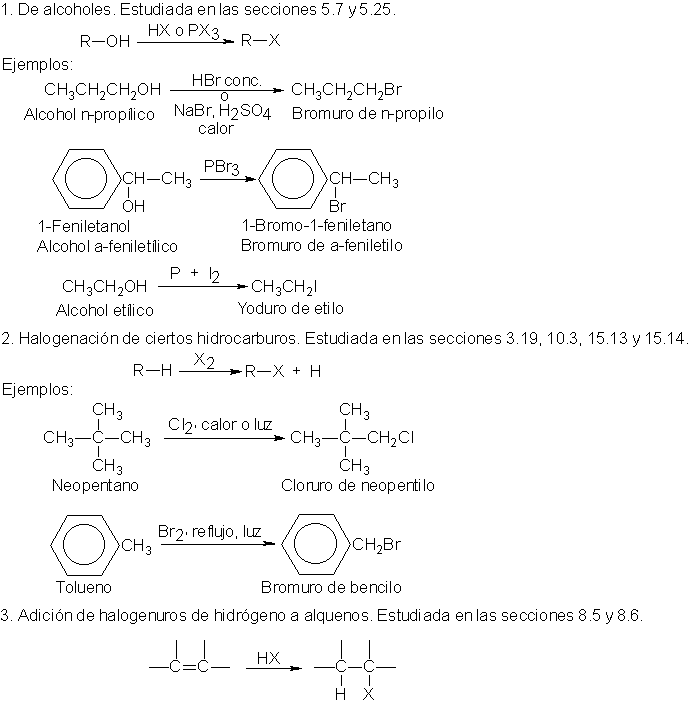

Los halogenuros de alquilo
se preparan casi siempre a partir de alcoholes.
A su vez, los alcoholes existen en una amplia variedad de formas y tamaños, y
los más simples se producen comercialmente (Sec. 17.6), mientras que los más
complicados se pueden sintetizar sin dificultades (Secs. 17.8, 17.14 y 17.15).
Aun cuando ciertos alcoholes tienden a reordenarse durante el reemplazo del OH
por X (Sec. 5.25), esta tendencia puede minimizarse empleando los halogenuros
del fósforo.
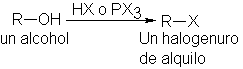
Para la síntesis de compuestos alifáticos en laboratorio, los
alcoholes son el punto de partida más común, siendo al conversión del alcohol
en un halogenuro de alquilo una de las primeras etapas más corrientes en tales
síntesis. Una vez obtenido el halogenuro de alquilo, la síntesis puede
continuar por decenas de vías, dependiendo de la reacción a la que se somete el
halogenuro, y como veremos en la sección siguiente, hay decenas de posibilidades.
Virtualmente nunca se preparan halogenuros de alquilo por halogenación directa de
alcanos. Desde el punto de vista de la
síntesis de laboratorio, un alcano es un punto muerto. La halogenación da,
por lo general, una mezcla de isómeros; aunque predomine en ocasiones un
isómero como en la bromación de isobutano, por ejemplo, probablemente se trate
del que no se desea. Cuánto más práctico es elegir un alcohol que tenga su OH
en la posición apropiada, y reemplazar luego este OH por un halogenuro.
A menudo se prepara un yoduro de sodio en acetona; se precipita el
bromuro o cloruro de sodio, menos solubles, de la solución y se puede eliminar
por filtración.
5.8 Reacciones de los halogenuros de alquilo.
Sustitución nucleofílica
alifática
Cuando se trata bromuro de metilo con hidróxido de sodio en un
disolvente capaz de disolver ambos reactivos,
se obtienen metanol y bromuro de sodio. Esta es una reacción de sustitución:
se sustituye el grupo OH por Br en el compuesto original.
![]()
El fenómeno es claramente heterolítico: el ión halogenuro saliente
lleva consigo al par electrónico que compartía con el carbono; el ión hidróxido
aporta el par de electrones necesario para la unión con el carbono. El carbono
pierde un par de electrones y gana otro. Este es sólo un ejemplo de la clase de
reacciones llamada sustitución
nucleofílica alifática.
La sustitución nucleofílica es característica de los halogenuros de
alquilo. Para entender por qué es así,
debemos fijar nuestra atención en el grupo funcional de esta familia: el
halógeno.
Un ión halogenuro es una base muy débil, lo que se refleja en su
disposición a ceder un protón a otras bases, es decir, en la gran acidez de los
halogenuros de hidrógeno. En un halogenuro de alquilo, el halógeno está unido a
un carbono y, al igual que el halogenuro libera con facilidad un protón,
también libera carbono nuevamente, hacia otars bases.
Estas bases poseen un par de electrones no compartido y buscan un
lugar relativamente positivo; es decir, buscan un núcleo con el cual compartir
su par electrónico.
Reactivos básicos, ricos en electrones, que tienden a atacar
el núcleo del carbono se conocen
como reactivos nucleofílicos
(del griego, <<que aman núcleos>>) o, simplemente, nucleófilos. Cuando este ataque termina
en sustitución, la reacción se denomina sustitución
nucleofílica.

El compuesto carbonado que sufre un tipo particular de reacción aquí,
el compuesto en el que tiene lugar la sustitución se llama sustrato. En el caso de la sustitución nucleofílica, el sustrato se
caracteriza por la presencia de un grupo
saliente: aquél que es desplazado del carbono y, llevando consigo el par de
electrones, se aleja de la molécula.
Debe tenerse presente que el nucleófilo :Z puede tener carga
negativa o ser neutro, y entonces el
producto R:Z será neutro o tendrá carga positiva. El sustrato R:W puede ser
neutro o positivo, y entonces el grupo saliente será negativo o neutro.
En el ejemplo inicial, el sustrato es el bromuro de metilo, el bromuro
es el grupo saliente y el ión hidróxido es el nucleófilo.
Debido a que el ión halogenuro débilmente básico es un buen grupo
saliente, los halogenuros de alquilo son excelentes sustratos para la
sustitución nucleofílica. Reaccionan con numerosos reactivos nucleofílicos,
tanto inorgánicos como orgánicos, para dar una amplia variedad de productos
importantes. Estos reactivos no sólo incluyen iones negativos, como hidróxido,
alcóxido o cianuro, sino también bases neutras, como amoniaco y agua; su
característica es la posesión de un par de electrones no compartido.
Como herramienta de síntesis, la sustitución nucleofílica es una de
las tres o cuatro clases más útiles de reacciones orgánicas. La sustitución
nucleofílica es el caballo de batalla de la síntesis orgánica; en sus diversas
formas, se trata de la reacción a la que recurriremos en primer luagr al
enfrentarnos con la tarea básica de reemplazar un grupo funcional por otro.
Dijmos que la síntesis de compuestos alifáticos comienza la mayoría de
las veces con alcoholes. Sin embargo,
descubriremos que el OH es un grupo saliente pobrísimo; solamente su
conversión en halogenuros de alquilo u
otros compuestos con buenos grupos salientes abre la puerta a la sustitución
nucleofílica.
Más adelante se da una larga de sustituciones nucleofílicas que indica
la versatilidad de los halogenuros de alquilo; muchas de ellas se dejarán para
ser tratadas en capítulos posteriores.
Con la sustitución nucleofílica descubriremos muchas cosas nuevas: una
reacción nueva, por supuesto de hecho, varias reacciones nuevas y una clase nueva de partícula reactiva, el carbocatión. Para descubrir lo que
sucede en estas reacciones, utilizaremos una herramienta nueva, la cinética, y otra antigua, la estereoquímica, en una forma nueva. Se
nos presentarán factores nuevos que afectan a la reactividad dispersión de carga, factores polares,
impedimento estérico, ayuda nucleofílica. Con ellos trabajaremos a lo largo
de nuestro estudio.

En el capítulo 6, continuando el estudio de la sustitución
nucleofílica, veremos cómo se afecta la reactividad con el disolvente, y con
ella, el curso de la reacción. Posteriormente, en el capítulo 20, utilizaremos
la sustitución nucleofílica como punto de partida para el estudio de sinforia el encuentro de los reactivos en la relación espacial
adecuada y veremos cómo podemos
controlar, en un grado antes posible, lo que sucede en una reacción química:
velocidad, orientación e incluso el resultado estereoquímico.
Los halogenuros de alquilo no sólo sufren sustitución, sino también eliminación, una reacción que
trataremos en el capítulo 7. Tanto la sustitución como la eliminación son
inducidas por medio de reactivos básicos, por lo que siempre habrá competencia entre ambas reacciones.
Estaremos interesados en dilucidar cómo se ve afectada esta competencia por
factores tales como la estructura del halogenuro y la naturaleza del nucleófilo
particular que se utilice como reactivo.
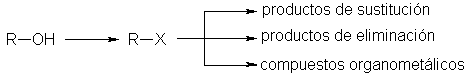
Los halogenuros de alquilo son las sustancias que más a menudo son convertidas en compuestos organometálicos:
compuestos que contienen carbono ligado a un metal magnesio (como en el
reactivo de Grignard), litio, cobre y
muchos otros. Ya nos hemos encontrado con algunos de estos compuestos y
seguiremos teniendo mucho que hacer con ellos a medida que vayamos avanzado. Veremos (Sec. 11.13)
que la conversión de halogenuros de alquilo en compuestos organometálicos
modifica la naturaleza del átomo de
carbono central de una manera fundamental, proporcionándonos una clase de
reactivos con propiedades únicas.
5.9 Sustitución nucleofílica alifática.
Nucleófilos y grupos
salientes
Los componentes requeridos para la sustitución nucleofílica son: sustrato, nucleófilo y disolvente. El sustrato
consta de dos partes: grupo alquilo y
grupo saliente. Nos ocuparemos del grupo alquilo durante gran parte de este
capítulo; estudiaremos las funciones del disolvente en el capítulo 6. Por el
momento examinaremos los otros componentes de estos sistemas: los nucleófilos y
grupos salientes.
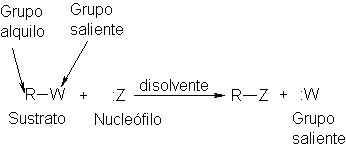
Ya hemos visto lo suficiente para darnos cuenta de que la basicidad
tiene un lugar importante en la comprensión de los nucleófilos y de los grupos
salientes. Los nucleófilos se caracterizan por ser bases, y los grupos
salientes, por ser bases débiles.
Podemos encontrar una correlación aproximada entre grado de basidad, por una parte,
y poder nucleófilo o capacidad de salida, por otra: entre dos bases, la
de mayor poder nucleofilico es, a menudo, la más fuerte, mientras que
corrientemente el mejor grupo saliente es la base más débil. Sin embargo, esto sólo se cumple para
conjuntos de nucleófilos o de grupos salientes estrechamente relacionados:
aquellos que, entre otras cosas,
implican un mecanismo elemento central oxígeno, por ejemplo, o
nitrógeno. Hay muchas excepciones a tales correlaciones, siendo, evidentemente,
la basicidad sólo uno de los factores
involucrados.
Debemos comprender con claridad la diferencia entre basicidad y poder nucleofílico o capacidad de salida.
Todas tienen que ver con la tendencia o, en el caso de la capacidad de salida,
con la falta de tendencia a compartir
un par de electrones para formar un enlace covalente. Sin embargo, hay dos diferencias
fundamentales:
(a) La basicidad es una cuestión de equilibrio;
el poder nucleofílico y la tendencia a salir son cuestiones de velocidad. De dos bases, se dice que una es más fuerte porque, en equilibrio,
fija una mayor proporción del ácido. De dos nucleófilos, el más poderoso es el
que ataca más velozmente al carbono;
de dos grupos salientes, se dice que uno es mejor que el otro, porque abandonada más velozmente al carbono.
(b) La basicidad (en el sentido de Lowry-Bronsted) implica una interacción
con un protón; el poder nucleofílico y la capacidad de salida implican
interacciones con el carbono.
Muchos de los productos que se
forman no son nuevos, pero por ahora sólo necesitamos apreciar cómo la
estructura de un producto en particular es el resultado natural de la
estructura de un determinado nucleófilo. Por el momento, utilizaremos
halogenuros de alquilo como ejemplos de sustratos.
Algunos nucleófilos son aniones, como el ión hidróxido;
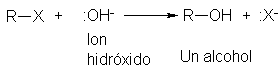
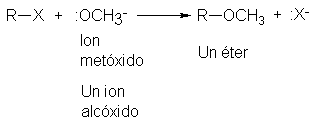
Los alcóxidos estrechamente
relacionados, como el metóxido;
Cianuro (el anión fuertemente básico de un ácido muy débil, HCN);

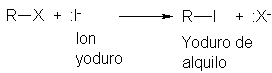
O incluso otro ión halogenuro que,
aun siendo sólo débilmente básico, tiene un par de electrones no compartidos:
Pero las moléculas neutras también pueden poseer electrones no
compartidos, ser básicas y, en consecuencia, actuar como nucleófilos. El agua, por ejemplo, es capaz de atacar
un halogenuro de alquilo para dar, finalmente, un alcohol. El oxígeno del agua
ya tiene dos hidrógenos, de manera que cuando se liga al carbono, inicialmente
no se forma el alcohol
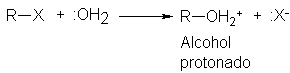
Mismo, sino ácido conjugado, es decir, el alcohol protonado; éste se transforma con facilidad en el alcohol por pérdida del protón.
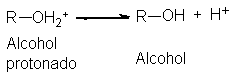
Aquí surge un aspecto importante. A menudo señalaremos por conveniencia la pérdida o ganancia de un ión
hidrógeno, H+, pero debe tenerse presente que no estamos realmente
en presencia de un protón desnudo, sino que estamos más bien transfiriendo un protón de una base a
otra. En este caso, por ejemplo, se convierte el alcohol protonado en alcohol
por la transferencia del protón al agua, casi tan básica como el propio alcohol
y mucho más abundante.
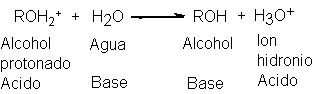
Observemos ahora algunos de los grupos
salientes que encontraremos. Hasta aquí hemos empleado como ejemplos
principales los halogenuros de alquilo y continuaremos haciéndolo en las
secciones siguientes. Sin embargo, debemos considerar que estas reacciones
tienen lugar exactamente de la misma forma con otros sustratos: compuestos que
contienen, al igual que los halogenuros, grupos salientes muy buenos.
De estos otros sustratos, en lugar de los halogenuros de alquilo se
utilizan más corrientemente los alquil ésteres de ácidos sulfónicos, ArSO2OR:
sobre todo en el estudio de mecanismos de reacciones, pero también en síntesis.
Los ácidos sulfónicos, ArSO3H, están relacionados con el ácido
sulfúrico y como éste son ácidos fuertes.

Sus aniones, los sulfonatos, son bases débiles y, en consecuencia,
grupos salientes buenos:
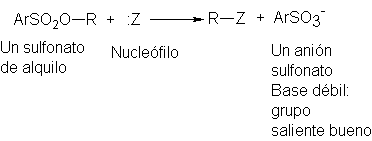
Los ésteres del ácido p-toluenosulfónico son los más empleados: los
p-toluenosulfonatos.
(Comprenderemos las estructuras de estos compuestos aromáticos más
adelante; por ahora basta con saber que se trata de grupos salientes buenos.)
El nombre del grupo p-toluenosulfonato suele abreviarse a tosilo (Ts), con lo que los p-toluenosulfonatos se convierten en tosilatos (TsOR).
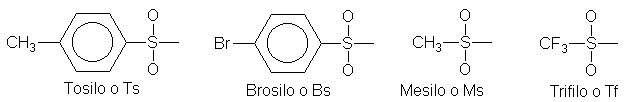
Al igual que los halogenuros de alquilo, los sulfonatos de alquilo
también se preparan con facilidad a partir de alcoholes. Por ejemplo:
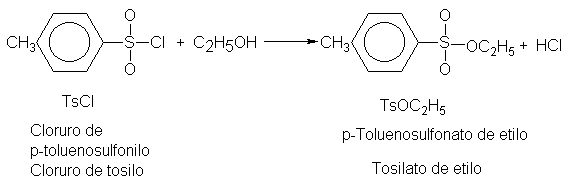
En la sección 18.5 veremos que las dos síntesis difieren en un aspecto
muy importante.
Comencemos ahora nuestro estudio del mecanismo de la sustitución
nucleofílica alifática. Esta reacción ha fascinado a los químicos durante
generaciones, incluidos muchos de los <<famosos>>, cuyos nombres
son o nos llegarán a ser familiares: J.A. LeBel, G.N. Lewis, T. M. Lowry; y
aquel gigante de la química orgánica, Emil Fischer, que abrió los dos campos
vastísimos de los hidratos de carbono y las proteínas.
En la actualidad, el área de la química orgánica más ampliamente
estudiada es la sustitución nucleofílica alifática en sus diversas formas y
también la más polémica. La fascinación y el argumento se centra en dos
cuestiones relacionadas. El enlace del grupo saliente se está destruyendo, y el
del nucleófilo se está formando. (a) ¿Cuál es la regulación en el tiempo de estos dos procesos? (b) ¿De dónde
procede al energía requerida para la ruptura del enlace con el grupo saliente?
Comenzaremos el estudio del mecanismo en el punto donde comienza la
historia moderna de la reacción: con la cinética
de la sustitución nucleofílica alifática. Pero, antes de nada, ¿qué es la
cinética?
5.10 Velocidad de reacción: efecto de la
concentración. Cinética
Hemos visto (Sec. 2.18) que la velocidad de una reacción química puede
expresarse como un producto de tres factores:
![]()
Hasta ahora hemos utilizado esta relación para comprar velocidades de
reacciones diferentes: para ayudarnos
a comprender la orientación y la reactividad relativa, y por qué tiene
lugar una reacción. Para que estas comparaciones
resulten lo más justas posibles, mantenemos
iguales aquellas condiciones que podemos controlar: temperatura y
concentración. Si se hace esto, entonces las reacciones íntimamente
relacionadas proceden a velocidad diferente, porque por lo general tienen
factores energéticos distintos; es decir, sus valores de Eact son
distintos. Para justificar esta diferencia, debemos estimar estabilidades
relativas de estados de transición.
También es útil el estudio de una reacción individual para ver cómo afectan los cambios deliberados de
condiciones experimentales a su velocidad. Por ejemplo, podemos determinarla
Eact si medimos la velocidad a diferentes temperaturas (Sec. 2.18). Sin
embargo, la información más valiosa que puede obtenerse acerca de una reacción
procede quizá del estudio del efecto de los cambios
de concentración sobre su velocidad.
A temperatura constante, ¿cómo se ve afecta la velocidad de una
reacción con un cambio en la concentración de los reaccionantes? Un aumento de
concentración no puede alterar la fracción de colisiones con energía suficiente
ni la fracción apropiada; sólo puede servir para aumentar el número total de
colisiones. Si se confinan más moléculas en el mecanismo espacio, chocarán con
más frecuencia y la reacción será más rápida. La frecuencia de colisiones y,
por tanto, la velocidad dependen con mucha exactitud de la concentración.
El campo de la química que se ocupa de la velocidad de las reacciones,
y especialmente la que depende de las concentraciones, se denomina cinética. Vemos qué nos puede enseñar
la cinética en cuanto a la sustitución nucleofílica alifática.
5.11 Cinética de la
sustitución nucleofílica alifática.
Reacciones de segundo y
primer orden
Tomemos como ejemplo específico la reacción del bromuro de metilo con
hidróxido de sodio
Para generar metanol:
![]()
Esta reacción probablemente se llevaría a cabo en etanol acuoso, en el
que ambos reactivos son solubles.
Si la reacción es el resultado de la colisión entre un ión hidróxido y
una molécula de bromuro de metilo, esperaríamos que la velocidad dependa de la
concentración de ambos reactivos. Si dobláremos la concentración de OH-,
[OH-], o la del CH3Br, [CH3Br], debería
duplicarse la frecuencia de colisiones
y en consecuencia, su velocidad. Si cualquiera de las concentraciones se reduce
a la mitad, también debería reducirse a
la mitad el número de colisiones y, por consiguiente, la velocidad.
Lo expuesto puede comprobarse experimentalmente. Se dice que la
velocidad de la reacción depende tanto de [OH-] como de CH3Br,
y viene expresada por
![]()
Si las concentraciones se expresan en moles por litro, por ejemplo,
entonces K es el número que, multiplicado por estas concentraciones, indica
cuántos moles de metanol se forman en cada litro por segundo. Para una temperatura
y un disolvente dados, k siempre tiene el mismo valor y es característico de
esta reacción específica; k se denomina constante
de velocidad. Así, para la reacción entre el bromuro de metilo y el ión
hidróxido en una mezcla de 80% de etanol y 20% de agua a 55ºC, el valor de k es
0.0214 litros por mol por segundo.
Por supuesto, no debe sorprendernos lo que se acaba de exponer.
Sabemos que un incremento de concentración ocasiona un aumento de la velocidad.
Pero veamos ahora la reacción correspondiente entre el bromuro de t-butilo y el
ión hidróxido:
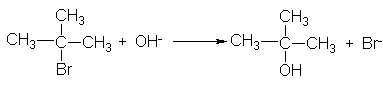
Si, como antes, duplicamos [RBr], también se duplica la velocidad; si
reducimos [RBr] a la mitad, decrece la velocidad en la misma proporción. En
cambio, si duplicamos [OH-] o lo reducimos a la mitad, no hay cambio
en la velocidad: la velocidad de la
reacción es independiente de [OH-].
La velocidad de la reacción del bromuro de t-butilo depende sólo
[RBr]. Esto se indica por la expresión
![]()
Para la reacción del bromuro de t-butilo en alcohol al 80% y a 55ºC,
la constante de velocidad es 0.010 por segundo, lo que significa que por cada
mol de bromuro de t-butilo presente, cada segundo reacciona 0.010 mol,
cualquiera que sea la concentración de [OH-].
Se dice que la reacción del bromuro de metilo sigue una cinética de segundo orden, puesto que
su velocidad depende de las concentraciones de dos sustancias. La del bromuro
de t-butilo, en cambio, sigue una cinética
de primer orden, pues su velocidad depende de la concentración de una sola sustancia.
5.12 Sustitución
nucleofílica alifática: dualidad de mecanismos
En la década de 1930 se realizaron estudios cinéticos de la
sustitución nucleofílica con diversos sustratos, detectándose los siguiente:
como el metilo, los sustratos primarios reaccionan con una cinética de segundo
orden, mientras que sustratos terciarios, como el t-butilo, lo hacen con una
cinética de primer orden. Los sustratos secundarios exhiben un comportamiento
intermedio: a veces de segundo orden; otras, de primer orden, y a menudo, una
mezcla de ambos.
Aparte del orden cinético, los estudios de la velocidad revelaron algo
más sobre la sustitución: las reactividades relativas de los diversos
sustratos. A una concentración dada de un nucleófilo, como el OH-,
se observó que la reactividad variaba aproximadamente como sigue:
![]()
Esto es, a medida que se avanza a lo largo de la serie CH3,
1º, 2º, 3º, la reactividad decrece en un comienzo, pasa por un mínimo (por lo
general en el 2º), y finalmente aumenta (véase Fig. 5.1). Es significativo que
el mínimo ocurre justamente en el punto de la serie donde la cinética cambia de
segundo a primer orden.
E. D. Hughes y sir
Chistopher (University College, Londres) utilizaron en 1935 estos dos conjuntos
de hechos orden cinético y reactividad relativa para elaborar una teoría amplia
de la sustitución nucleofílica alifática. La piedra angular de su teoría fue
que la sustitución nucleofílica alifática
puede proceder por dos mecanismos distintos, a los que denominaron, por
razones que se aclaran luego, SN2 y SN1. Sustratos
diferentes reaccionan con órdenes cinéticos distintos porque están reaccionando
por mecanismos diferentes: algunos, como el metilo, por el SN2;
otros, como el t-butilo, por el SN1.
Debido a que el mecanismo cambia en este punto, de SN2
a SN1, la reactividad pasa
por un mínimo con sustratos secundarios. A medida que se procede a lo largo de
una serie lógica, la aparición de un mínimo o un máximo en una propiedad
reactividad, acidez, actividad antibacteriana sugiere que operan dos factores
opuestos. Para este caso, Hughes e Ingold propusieron que los factores serían
las secuencias de reactividad opuestas en ambos mecanismos distintos. A medida
que se avanza en la serie, decrece la reactividad del mecanismo SN2
de CH3 a 1º, y en 2º es tan baja que comienza a contribuir la
reacción SN1 de forma significativa; la reactividad aumenta fuertemente, ahora
por SN1, a 3º (Fig. 5.1).

En las siguientes secciones, veremos lo que son estos dos mecanismos,
los hechos en los que se basan y cómo explican estos hechos. Por ejemplo,
veremos como explican la diferencia en el orden cinético y, en particular, el
hecho desconcertante de que la velocidad de la reacción del bromuro de t-butilo
es independiente de la [OH-]. Estudiaremos los factores considerados
como responsables del esquema de las secuencias de reactividad opuesta para los
dos mecanismos. Finalmente, veremos cómo este patrón de mecanismo diseñado en
1935 ha resistido la prueba del tiempo.
5.13 Reacción SN2: mecanismo y cinética
La reacción entre el bromuro de metilo y el ión hidróxido para dar
metanol sigue una cinética de segundo orden; es decir, su velocidad depende de
ambos reactivos:
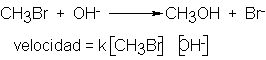
La forma más simple de definir
la cinética es suponer que la reacción requiere una colisión entre un ión
hidróxido y una molécula de bromuro de metilo. Por las evidencias que
estudiaremos en breve, se sabe que el ión hidróxido permanece lo más alejado
posible del bromo durante el ataque, o sea, el ión ataca a la molécula por
atrás.
Se cree que la reacción sucede como se indica en la figura 5.2 Cuando
el ión hidróxido choca con una molécula de bromuro de metilo en la cara más
remota del bromo, y con energía suficiente, se forma un enlace C-OH y se rompe
el enlace C-Br, liberándose el ión bromuro.
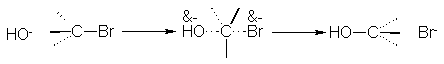
Fig. 5.2 Reacción SN2: inversión completa de la
configuración. El reactivo
Nucleofílico ataca la parte de atrás.
El estado de transición puede representarse como una estructura en la que
el carbono está parcialmente unido al –OH y al –Br; aún no se ha formado por
completo el enlace C-OH, ni se ha roto
en su totalidad el enlace C-Br. Ha disminuido la carga negativa del hidróxido,
porque ha comenzado a compartir sus electrones con el carbono. El bromo ha
comenzado a desarrollar una carga negativa parcial, ya que ha eliminado en
parte un par de electrones del carbono.
El –OH y el –Br se ubican lo más apartados posibles: los tres hidrógenos y el carbono se encuentran en un
solo plano, y todos sus ángulos de enlace son de 120º. En consecuencia, los
enlaces C-H se distribuyen como los rayos de una rueda, con los enlaces C-OH y
C-Br ubicados a lo largo de su eje.
Podemos observar cómo se alcanza la geometría del estado de
transición. El carbono sujeta a los tres hidrógenos por medio del solapamiento
de tres orbitales sp2, que son trigonales y, por tanto, planos, y
están alejados 120º. Los enlaces parciales con el grupo saliente y con el
nucleófilo se forman por el solapamiento de vestigios de los orbitales p,
restantes, separados 180º y perpendiculares al plano de los orbitales sp2.
Este es el mecanismo denominado SN2: sustitución nucleofílica bimolecular. Se emplea aquí el término
bimolecular, porque la etapa que determina la velocidad implica la colisión de
dos partículas.
¿Qué evidencia hay de que los halogenuros de alquilo reaccionen de
esta manera? En primer lugar, acabamos de ver que este mecanismo es compatible
con una cinética de reacción como la del bromuro de metilo con ión hidróxido.
En general, una reacción SN2
sigue una cinética de segundo orden. Analicemos otras evidencias.
5.14 Reacción SN2:
estereoquímica, Inversión de la configuración
Tanto el 2-bromooctano como el 2-octanol son quirales; es decir, tienen moléculas que no pueden suponerse
a sus imágenes especulares. En consecuencia, estos compuestos pueden existir
como enantiómeros y ser ópticamente activos. Se ha obtenido el 2-octanol
ópticamente activo por resolución de la modificación racémica (Sec. 4.27), y a
partir de él se ha sintetizado el 2-bromooctano activo.
Se han asigando las configuraciones siguientes (Sec. 4.24):

Observamos que el (-)-bromuro y el (-)-alcohol tienen configuraciones similares,
es decir, el OH ocupa en el (-)-alcohol la misma posición relativa que el –Br
en el (-)-bromuro. Como sabemos, los compuestos de configuración similar no necesariamente rotan la luz en la
misma dirección; en este caso, ocurre que lo hacen. [También sabemos que
compuestos de configuración similar no tienen por qué recibir la misma
especificación R o S (Sec. 4.24); es coincidencia, nuevamente, que en este caso
ambos sean R.]
Al hacer reaccionar (-)-2-bromooctano con hidróxido de sodio en
condiciones que favorecen una cinética de segundo orden, se obtiene
(+)-2-octanol.
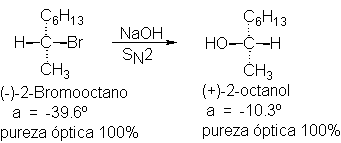
Observamos que el grupo –OH no ha tomado la posición ocupada con
anterioridad por el Br; el alcohol
tiene una configuración opuesta a la del bromuro. Una reacción que da un producto con configuración opuesta a la del
reactivo procede con inversión de la
configuración.
(En este caso particular, la inversión de la configuración va
acompañada, casualmente, de un cambio de especificación, de R a A, pero esto no
siempre es así. No podemos determinar si una reacción procede con inversión o
retención de configuración inspeccionando simplemente las letras empleadas para
especificar al reactivo y al producto: debemos establecer y comprar las
configuraciones absolutas indicadas por tales letras.)
Surge ahora la pregunta: ¿procede una reacción como ésta con inversión
completa?
Es decir, ¿se invierte la configuración de todas las moléculas? La
respuesta es sí. Una reacción SN2 procede con inversión estereoquímica
completa.
Para responder a una pregunta como ésta, debemos conocer la pureza
óptica del reactivo del que partimos y del producto que obtenemos; en este
caso, el 2-bromooctano y el 2-octanol. Para conocer éstos, a su vez, debemos
saber cuál es la rotación máxima del bromuro y la del alcohol; es decir,
debemos conocer la rotación de una muestra ópticamente pura de cada uno de
ellos.
Por ejemplo, supongamos que sabemos que la rotación máxima del
2-bromooctano ópticamente puro es 39.6º y que la del 2-octanol ópticamente
puro, sabemos que la reacción procedió con inversión completa. O bien y esto es
más práctico, si una muestra del halogenuro de rotación –32.9º, por ejemplo
(pureza óptica 83%), diera alcohol de rotación +8.55º (pureza óptica 83%),
también sacaríamos la misma conclusión.
Durante el desarrollo de las ideas de las reacciones SN1 y
SN2, Hughes e Ingold estudiaron la reacción del 2-bromooctano
ópticamente activo y obtuvieron resultados que les llevaron a concluir que la
reacción SN2 procede, dentro de los límites del error experimental,
con inversión completa.
El valor específico que Hughes e Ingold utilizaron para la rotación
del 2-bromooctano ópticamente puro ha sido puesto en duda, pero la idea básica
de la inversión completa en reacciones SN2 se ha establecido más
allá de toda duda: por medio del estudio de sistemas distintos de los
halogenuros de alquilo y por medio de un elegante trabajo sobre radiactividad y
actividad óptica (véase Problema 5.5).
El ataque por atrás en la sustitución del tipo SN2 se
propuso originalmente para justificar
la inversión de la configuración. A medida que el OH se va uniendo al carbono,
tres enlaces se ven forzados a separarse hasta alcanzar la disposición radial
del estado de transición; luego, al expulsarse el bromuro, siguen desplazándose
hasta alcanzar una disposición tetraédrica opuesta
a la original. Este proceso se compara a menudo con el paraguas que se da vuelta en un temporal.
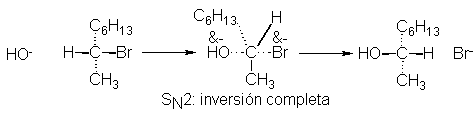
La estereoquímica de la reacción del 2-bromooctano indica ataque por atrás
de acuerdo con el mecanismo SN2; estudio de otros compuestos ópticamente
activos, en condiciones en las que las reacciones siguen una cinética de
segundo orden, muestran resultados similares. No es posible estudiar la
estereoquímica de la mayoría de los halogenuros, puesto que no son ópticamente
activos, sin embargo no hay razón para dudar que también ellos sufren ataque
por atrás.
La inversión de la configuración es la regla general para reacciones
que suceden en centros quirales, y es mucho más común que la retención de la
configuración. Extrañamente, la frecuencia de la inversión fue la causa de su
difícil descubrimiento. Paul Walden (Politécnico de Riga, Letonia) descubrió el
fenómeno de la inversión en 1896, cuando encontró una de las reacciones excepcionales,
en la cual la inversión no sucede.
Aparte de la orientación espacial del ataque, hay otra característica
de la sustitución SN2 aún más fundamental, puesto que define el
mecanismo: la reacción sucede en una sola
etapa, de modo que la ruptura y formación de enlaces ocurren
simultáneamente, de una manera concertada.
También esta característica se apoya en la estereoquímica: no por el hecho de
que hay inversión, sino porque ésta es completa.
Cada molécula del sustrato sufre el mismo destino estereoquímico inversión,
como es el caso. Esta especificidad es consistente con el mecanismo: el grupo
saliente todavía se encuentra ligado al carbono cuando comienza el ataque
nucleofílico, controlado así la dirección del ataque. (Apreciaremos mejor la
importancia de este aspecto al ver el contraste que ofrece la reacción SN1.)
De hecho, ya hemos encontrado una situación de contraste: la cloración por radicales libres
1-cloro-2-metilbutano ópticamente activo (Sec. 4.28). Se extrae primero
hidrógeno del centro quiral; luego, en un paso posterior, se une cloro al
carbono. Sin embargo, una vez que el hidrógeno se fue, no queda nada que pueda
dirigir el cloro hacia una cara en particular del carbono: el ataque ocurre al
azar en ambas caras, de modo que resulta la modificación racémica.
En consecuencia, el mecanismo SN2 está apoyado en pruebas
estereoquímicas. De hecho, la relación entre mecanismo y estereoquímica está
tan bien establecida que, en ausencia de otra evidencia, se considera que una
inversión completa indica una reacción SN2.
Podemos apreciar, una vez más,
que la estereoquímica puede proporcionar un tipo de información acerca
de una reacción que no es posible obtener
por otros medios.
5.15 Reacción SN2: reactividad. Impedimento estérico
Examinemos ahora la reactividad
en la sustitución nucleofílica alifática, y veamos cómo la afectan los cambios
estructurales del grupo alquilo.
De acuerdo con la teoría del mecanismo en 2º, es sencillamente la mezcla
de dos órdenes de reactividad opuestos: uno por SN2, y el otro por SN1.
Una manera de poner a prueba esta hipótesis es efectuado una sustitución en
condiciones tales que todos los miembros de una serie desde metilo hasta 3º
reaccionen en gran medida mediante una cinética de segundo orden, y midiendo
las constantes de velocidad de segundo orden; a continuación, se repite todo el
proceso, seleccionando esta vez condiciones que favorezcan una reacción de
primer orden, con la correspondiente determinación de las constantes de
velocidad de primer orden.
Observemos resultados obtenidos de esta manera: primero, para la
reacción SN2, y más adelante, en otra sección, para la reacción SN1.
Las mediciones directas de velocidades SN2 para una serie
de sustratos detectan resultados como los siguientes. (DMF es dimetilformamida, un disolvente que
favorece la reacción SN2, como veremos en la Sec. 6.4.)
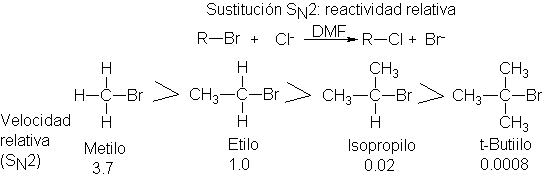
Según lo postulado, la reactividad de los sustratos en la reacción SN2
es:
![]()
¿Cómo podemos justificar este orden de reactividades? Para responder a
una pregunta de este tipo, debemos observar la reacción específicamente implica
en este caso, la reacción SN2 y comparar la estructura de los
reactivos con la del estado de transición esta vez no es intermediaria entre
las de los reactivos y la del producto; esta vez, no podemos esperar que los
factores que estabilizan el producto también lo haga con el estado de
transición.
Durante el desarrollo de muchas reacciones, se produce un cambio en la
distribución electrónica tal que se desarrolla una carga negativa o positiva en
la molécula acomodada esa carga.
A su vez, el acomodo de la carga depende de los efectos polares de los sustituyentes, es decir, de lo efectivos que
sean estos sustituyentes para quitar o
proporcionar electrones. Por tanto, examinemos en primer lugar la reacción SN2
con respecto a cambios en la distribución de los electrones, utilizando
nuevamente como ejemplo la reacción de un halogenuro de alquilo con el ión
hidróxido. Tal como se describió el estado de transición
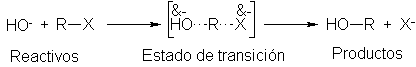
(Sec. 5-13), hay un enlace formado parcialmente entre el carbono y el
ión hidróxido, y otro parcialmente roto entre el carbono y el halógeno; el ión hidróxido
aportó electrones al carbono, mientras que el ión halogenuro se los quitó.
Salvo que alguno de estos procesos, la
formación o la ruptura del enlace, haya progresado mucho más que el otro, el
carbono no es apreciablemente más negativo o positivo que al inicio de la
reacción. Si fuera éste el caso, se concluiría que la secuencia de
reactividades para la reacción SN2 no es el resultado de efectos
polares de los grupos sustituyentes.
Para comprender la influencia de la estructura en la velocidad, comparemos
el estado de transición y los reactivos con respecto a sus formas, comenzando con la reacción del bromuro de metilo. El
carbono en el reactivo y en el producto es tetraédrico, mientras que en el
estado de transición se halla unido a cinco átomos. Como ya se indicó
anteriormente, los enlaces C-H presentan una disposición similar a la de los
radios de una rueda, mientras que lso enlaces C-OH y C-Br están en el eje (Fig.
5.3).
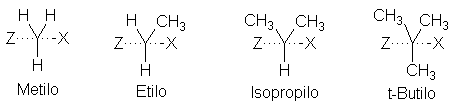
Fig. 5.3 Estructura molecular y reactividad: el factor estérico en la
reacción SN2. La aglomeración eleva la energía del estado de transición y reduce la velocidad de la
reacción.
¿Cuál sería el efecto de reemplazar sucesivamente los hidrógenos por
grupos metilo?
Esto es, ¿cómo diferiría el estado de transición, a medida que
avancemos, del bromuro de metilo al de etilo, luego al de isopropilo, y
finalmente al bromuro de t-butilo? Con el reemplazo paulatino de átomos de
hidrógeno por grupos metilo más voluminosos, va produciéndose una aglomeración
creciente en torno al carbono. Como puede apreciarse con los modelos a escala
(Fig. 5.4), cada vez resulta más inaccesible la parte de atrás de la molécula,
donde debe realizarse el ataque.
Esta aglomeración es particularmente severa en el estado de
transición, en el cual los metilos son forzados a acercarse al OH y al Br (Fig.
5.3). Las interacciones sin enlaces elevan la energía del estado de transición
aglomerado más que la energía del reactivo, menos congestionado: la Eact es más
elevada, y la reacción, más lenta.
Esta interpretación es la que se acepta generalmente ahora: las diferencias en la velocidad de dos
reacciones SN2 se deben principalmente a factores estéricos, y no a factores polares; es decir, las
diferencias de velocidad tienen relación con el volumen de los sustituyentes, y no con sus efectos sobre la
distribución electrónica. A medida que aumenta el número de sustituyentes
unidos al carbono portador del halógeno, disminuye la reactividad con respecto
a la sustitución SN2, como lo demuestran las mediciones para la
serie metilo, 1º, 2º, 3º.
Que aquí están operando factores estéricos se ve confirmado por las
velocidades relativas de otra serie de sustratos. Esta vez, todos los sustratos
son primarios, por lo que tienen el mismo número
de sustituyentes uno unidos al carbono que tiene el halógeno.
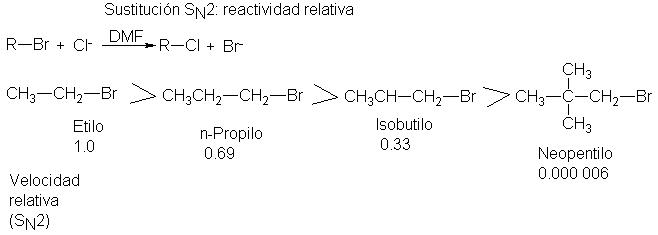
Pero ahora crece constantemente el tamaño
del sustituyente: en el bromuro de etilo, éste es metilo; en el bromuro de
n-propilo, etilo; en el bromuro de isobutilo, isopropilo, y en el bromuro de neopentilo,
el sustituyente es t-butilo. Y a medida que crece el tamaño del sustituyente
(único) aumenta también el impedimento estérico al ataque, de modo que decae la
velocidad. (Véase Fig. 5.5.)
Observamos entonces que el mecanismo SN2 recibe apoyo mediante
tres líneas de evidencia : cinética, estereoquímica y el efecto de la
estructura sobre la reactividad.
Retornaremos más adelante a la reacción SN2, pero por el
momento trataremos otro mecanismo que puede realizar la sustitución
nucleofílica alifática.
5.16 Reacción SN1: mecanismo y cinética.
Etapa determinante de la
velocidad
La reacción entre el bromuro de t-butilo y el ión hidróxido para dar
alcohol t-butílico sigue una cinética de primer orden; es decir, la velocidad
depende de la concentración de uno solo de los reactivos: el bromuro de
t-butilo.
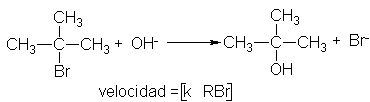
¿Cómo hemos de interpretar el hecho que la velocidad es independiente
de [OH-]? Si la velocidad de la reacción es independiente de [OH-],
sólo puede significar que la reacción, cuya
velocidad estamos determinando, no involucra OH-
Estas observaciones pueden interpretarse por el siguiente mecanismo:
el bromuro de t-butilo se disocia lentamente (paso 1) en un ión bromuro y en un
catión derivado del grupo t-butilo: un carbocatión.
Este carbocatión se combina ahora (paso 2) velozmente con un ión hidróxido para
generar alcohol t-butílico.
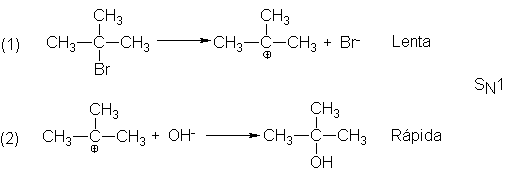
La velocidad de la reacción total queda determinada por la ruptura
lenta del enlace C-Br, para formar el carbocatión, el cual, una vez formado,
reacciona de inmediato para dar el producto. La velocidad que estamos
determinando corresponde a la del paso (1), que no involucra OH-, de
modo que no depende de [OH-]. Una
sola etapa cuya velocidad determinada la velocidad total de un proceso de varias
etapas se denomina etapa
determinante de la velocidad.
No es extraño que el paso que determina la velocidad sea el que
implica la ruptura de un enlace, un proceso que consume energía. Reconocemos
esta ruptura de enlace particular como un ejemplo de heterólisis, escisión en la que ambos electrones enlazantes parten
con el mismo fragmento: un proceso que necesita aún más energía (Sec. 1.14) que
la homólisis que estudiamos en la sustitución por radicales libres. (En la Sec.
6.5 descubriremos de dónde procede toda esta energía.)
Es evidente también que la combinación del carbocatión con el ión
hidróxido es un paso sumamente rápido, puesto que solamente implica la formación de un enlace, un proceso
liberador de energía. Esta combinación puede reconocerse como una reacción
ácido-base en el sentido de Lewis. Sabemos que el ión OH- es una
base fuerte; como veremos, los carbocationes son ácidos de Lewis fuertes.
Este es el mecanismo llamado SN1: sustitución nucleofílica unimolecular. Debido a que la etapa
determinante de la velocidad sólo depende de una molécula, empleamos aquí el
término unimolecular (sin considerar
las muchas moléculas necesarias de disolvente).
No debemos imaginar que las leyes de la química quedan mágicamente
suspendidas para el segundo paso, el rápido. Implica una reacción con OH-
y su velocidad depende de [OH-]. Lo especial aquí es que aumente el
paso (2) se haga más lento por una baja [OH-], sigue siendo mucho más rápido que el paso (1), por lo
que cualquier cambio en su velocidad no afecta la velocidad global.
Veamos lo que queremos decir con etapa determinante de la velocidad
para una reacción como la siguiente,
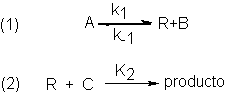
Donde R es un intermedio reactivo (carbocatión, radical libre, carbanión)
cuya concentración se mantiene en un estado
estacionario bajo durante toda la reacción. La expresión cinética para la
formación del producto es
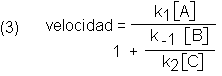
Sin entrar en la derivación de esta expresión, veamos su significado.
El término k1 [A] está en el numerador, y el k2
[C], en el denominador del dominador; cuanto más grandes son mayor es la velocidad. Esto es
razonable, puesto que k1 [A] es la velocidad del paso (1) y k2
[C] contribuye a la velocidad del paso (2). El término k-1 [B] se
encuentra en el denominador; cuanto mayor es, más lenta es la reacción: esto
también es comprensible, puesto que contribuye a la velocidad del reverso del
paso (1).
Ahora, si resulta que k2 [C] es mucho más grande que k-1 [B], entonces, el término k-1
[B]/k2 [C] es muy pequeño insignificante con respecto a 1-, por lo
que puede despreciarse; en tal caso, resulta la expresión conocida para una
cinética de primer orden:
![]()
Pero si K2 [C] es mucho mayor que k-1 [B],
significa que el paso (2) es mucho más
rápido que el reverso del paso (1). Esta es la verdadera condición para que
el paso (1) sea determinante de la velocidad. ¿Significa lo anterior,
contrariamente a lo dicho, que el paso (1) en su forma directa no necesita ser
más lento que el (2)? El paso (1) debe seguir siendo lento, pues de lo
contrario el intermedio reactivo se generaría más rápidamente de lo que se podría consumir, de modo que su
concentración se iría incrementando, lo que es contrario a la naturaleza del
intermediario reactivo, y es una condición diferente a quélla para la cual vale
la expresión cinética (3).
¿Qué pruebas hay de que los halogenuros de alquilo pueden reaccionar
por medio de este mecanismo? Acabamos de ver que el mecanismo concuerda con la
cinética de primer orden de una reacción como la del bromuro de t-butilo con el
ión hidróxido. En general, una reacción
SN1 sigue una cinética de primer orden. La velocidad de la
reacción completa queda determinada por la velocidad de ionización del
halogenuro de alquilo, de modo que sólo depende de su concentración.
En las siguientes secciones se vería otras evidencias, pero para
entenderlas debemos saber algo más acerca del intermedio que se encuentra en el
meollo del mecanismo y de hecho en el centro de gran parte de la química
orgánica, el carbocatión. Así pues,
dedicaremos un tiempo a seguir dos hebras que se entrelazan por el entramado de la química orgánica:
concentrándonos alternativamente en la sección SN1 y en la química
fundamental de los carbocationes.
Con el fin de explicar los hechos observados, se propuso cierto
mecanismo para la halogenación de los alcanos. El centro de este mecanismo es
la existencia efímera de radicales libres, partículas neutras altamente
reactivas que tienen un electrón no apareado.
Para describir ciertas observaciones de las cuales la cinética de
primer orden tratada en la sección anterior es tan sólo una, se ha propuesto
otra partícula: el carbocatión, un grupo de átomos que contiene un carbono
con seis electrones solamente.
Los carbocationes se clasifican como primarios, secundarios o
terciarios, de acuerdo con el carbono portador de la carga positiva. Se nombran
utilizando la palabra catión.
Por ejemplo:
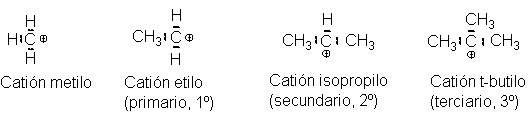
Debemos estas preparados para encontrarnos con otros dos nombres para
lo que hemos denominado carbocatión. Ión carbonio es casi el único nombre
utilizado en la literatura antigua, siendo aún su empleo muy común, aunque a
veces con un significado especial. Olah (más adelante) ha propuesto que se use
ión carbenio para las especies recién descritas, reservando ión carbonio para especies como el CH5+
(análogas al ión amonio, etc.); el
conjunto de iones carbenio y carbono serían los carbocationes.
AL igual que el radical libre, el carbocation es una partícula muy
reactiva, y por la misma razón: la tendencia a completar el octeto del carbono.
Puesto que en este caso se necesita un par de electrones para completarlo, el
carbono es un ácido de Lewis, y sumamente poderoso. Se distingue del radical
libre por poseer una carga positiva.
Un tipo de carbocatión excepcionalmente estable fue reconocido en 1902
(Problema 15.10, Sec. 15.17) debido al carácter salino de ciertos compuestos
orgánicos. No obstante, para cationes alquilo simple, tal observación directa
debe ser muy difícil, debido precisamente a la reactividad y de ahí su corta
vida que se les atribuye. Sin embargo, durante las décadas del 1920 y 1930 se
propusieron cationes alquilo como intermediarios en muchas reacciones orgánicas, aceptándose su existencia debido
principalmente al trabajo de tres químicos: Hans Meerwein, de Alemania, el
<<padre de la química moderna del ión carbonio>>; sir Christopher
Ingold, de Inglaterra, y Frank Whitmore, de Estados Unidos. La evidencia
consistió en una amplia gama de observaciones que se hicieron durante el
estudio de la química de halogenuros de alquilo, alcoholes, alquenos y muchos
otros tipos de compuestos orgánicos: observaciones que revelaron un patrón
básicamente similar de comportamiento, atribuido por lógica a carbocationes
intermediarios. Una parte importante de este libro se dedica al estudio de ese
patrón.
En 1963, George Olah (ahora en la Universidad de California del Sur)
informó sobre la observación directa
de cationes alquilo simples. Disueltos en el fuertísimo ácido de Lewis SbF5,
se observó que los fluoruros (y, más tarde, otros halogenuros) de alquilo
sufren ionización con formación de un catión, que se estudia con comodidad. Se
observó un cambio drástico en el espectro RMN (Cap. 16), del espectro del fluoruro
de alquilo al de una molécula que no contiene flúor, sino carbono híbrido sp2
con una densidad electrónica muy baja.
![]()
La figura 5.6 ilustra lo que se observó para el sistema del fluoruro de
t-butilo: un espectro sencillo, pero muy significativo precisamente por su
sencillez. Aunque potencialmente muy reactivo, el catión t-butilo tiene muy
poco que hacer en este ambiente, excepto tratar de recuperar el ión fluoruro y
el SbF5 es un ácido de Lewis aún más fuerte que el catión. Esta es
una reacción ácido-base; el SbF5 (llamado superácido) es un ácido de Lewis aun más fuerte que el catión
alquilo, y mantiene la base ganada, el ión fluoruro.
Por métodos como este, Olah abrió la puerta al estudio no sólo de la
propia existencia de cationes orgánicos de muchos tipos, sino también, de los
detalles íntimos de su estructura.
Puede llevarse a cabo un efecto muy significativo para esta reacción.
Si se diluye la solución que contiene el R+SbF6- con agua, se
obtiene el alcohol, ROH. En esencia con esto tenemos los dos pasos propuestos
para la reacción SN1 la generación de un carbocatión y su
combinación con un nucleófilo, pero observados como procesos discretos,
separados todo el tiempo que deseemos.
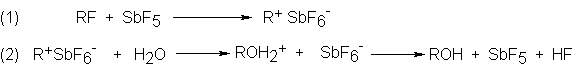
Por tener solamente un sexteto de electrones en su carbono, un
carbocatión es una partícula inestable y altamente reactiva. Como veremos,
puede sufrir una amplia gama de reacciones. Cuál de ellas depende de las
condiciones experimentales. Pero todas las reacciones de un carbocatión tienen
un fin común: proporcionar un par de
electrones para completar el octeto del carbono con carga positiva. En la
segunda etapa de una reacción SN1 probablemente vemos la manera más
directa de lograr esto: la combinación con un nucleófilo, una molécula básica
rica en electrones.

5.18 Estructura de carbocationes
En un carbocatión, el carbono deficiente en electrones se halla
enlazado a otros tres átomos mediante orbitales sp2. Estos orbitales
se encuentran en un plano (Sec. 1.10), el del núcleo del carbono, y están
dirigidos hacia los vértices de un
triángulo equilátero. Esta parte del carbocatión es, en consecuencia, plana, con el carbono deficiente
electrónicamente y los tres átomos unidos a él ubicados en el mismo plano (Fig.
5.7a).
Sin embargo, la descripción de la molécula aún no está completa. El
carbono todavía posee un orbital p, con sus dos lóbulos ubicados por encima y
por debajo del plano de los enlaces o (Fig. 5.7b); en un carbocatión, este
orbital p se encuentra vacío. A pesar
de estar formalmente vacío, descubriremos que este orbital p está íntimamente
implicado en la química de los carbocationes: en sus estabilidades y en las
estabilidades de los diversos estados de transición que conducen a su formación. Este es el resultado del
solapamiento del orbital p con
otros orbitales cercanos solapamiento
geométricamente posible por la planaridad del carbocatión.

No cabe duda acerca de la planaridad de los carbocationes. La descripción
mecanico-cuántica de un carbocatión es exactamente la misma que la del
trifluoruro de boro (Sección 1.10), una molécula cuya planaridad está
firmemente establecida. Los espectros
RMN e infrarrojos de los carbocationes estabilizados, estudiados por Olah, son
consistentes con la hibridación sp2 y la planaridad: especialmente
los espectros infrarrojo y Raman del catión t-butilo son sorprendentemente
similares a los del trimetilboro, el cual se sabe que es plano.
5.19 Reacción SN1:
estereoquímica
Seguiremos con la química fundamental de los carbocationes en la
sección 5.20, pero por ahora continuaremos con nuestro planteamiento original,
la sustitución nucleofílica. Veamos un aspecto directamente relacionado con la
forma de los carbocationes: la estereoquímica de la reacción SN1.
Según los estudios estereoquímicos de
la reacción SN2 (Sec. 5.14), la sustitución se efectúa en un
sustrato ópticamente activo. Se aísla el producto y se comparan su
configuración y pureza óptica con las del material de partida. Como antes,
deben haberse asignado las configuraciones relativas del reactivo y del
producto, y se deben conocer también las rotaciones de las muestras ópticamente
puras, para poder calcular las purezas ópticas.
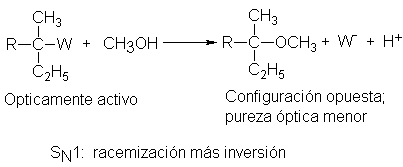
Se ha hecho este tipo de estudios de las reacciones entre varios
sustratos terciarios y el disolvente metanol, CH3OH: reacciones de
un tipo que con toda probabilidad proceden mediante SN1. En cada
caso se obtiene un producto de configuración opuesta de la del material de
partida y de pureza óptica
considerablemente inferior . Un sustrato ópticamente puro da, por ejemplo,
un producto cuya pureza óptica es de sólo el 50% y, en muchos casos, todavía
menor.
El material de partida ópticamente puro sólo contiene uno de los enantiómeros,
mientras que el producto debe contener los dos. El producto es así una mezcla
del compuesto invertido y de la modificación racémica, por lo que se dice que
la reacción procede con inversión más racemización
parcial.
Aclararemos bien lo anterior. Consideremos el caso en que un sustrato
ópticamente puro que da un producto con la configuración opuesta y de una
pureza óptica del 50%. De cada 100 moléculas de producto, se forman 75 con
inversión de la configuración y 25, con retención. Las 25 de configuraciónretenida
anulan la rotación de 25 moléculas de configuración invertida, con lo que queda
un exceso de 50 moléculas con configuración invertida para proporcionar la
rotación óptica observada: el 50% del valor máximo.
Podría decirse que la reacción procede con 75% de inversión y 25% de
retención; de igual manera, podría argumentarse que la reacción procede con 50%
de inversión y 50% de racemización. Sin embargo, esta última forma es la que
generalmente se utiliza: el porcentaje de racemización es, como veremos, una
medida de la aleatoriedad estereoquímica, y el porcentaje de inversión neta (o,
como sucede en ciertos tipos de reacciones, retención neta) es una medida de la
estereoselectividad (Sec. 9.2).
¿Cómo explicamos la estereoquímica observada para la reacción SN1?
Estudiaremos primero por qué sucede la racemización, y luego, por qué es sólo
parcial y va acompañada de alguna inversión neta.
En una reacción SN2 vimos que el nucleófilo ataca a la
propia molécula de sustrato (sección 5.14), y la inversión completa que se observa es una consecuencia directa de tal hecho:
el grupo saliente aun se encuentra ligado al carbono en el instante del ataque,
y dirige el ataque sobre cada molécula de la misma manera hacia la parte de
atrás. Ahora bien, en una reacción SN1, el nucleófilo no ataca al
sustrato, sino al intermediario, el carbocatión: el grupo saliente se ha
desprendido ya, y podría pensarse que no afecta a la orientación espacial del
ataque.
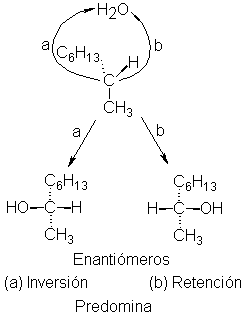
Veamos aqué conduce este razonamiento. El sustrato ópticamente activo
digamos, un halogenuro de alquilo se disocia en el primer paso para formar un
ión halogenuro y un carbocatión. Luego, el reactivo nucleófilo, Z:, se une al
carbocatión. Sin embargo, puede unirse a cualquiera de las caras de este ión
plano y, dependiendo de la cara que elija, da origen a uno u otro de los
productos enantiómeros (véase Fig. 5.8). Los dos enantiómeros juntos
constituyen la modificación racémica. Así, la racemización que acompaña a estas
reacciones es consistente con el mecanismo SN1 y con la formación de
un carbocatión intermedio.
[Hasta aquí, nuestro análisis de acuerdo con lo dicho sobre la
estereoquímica de la cloración por radicales libres (Sec. 4.28), en la que el
ataque aleatorio sobre ambas caras del radical
libre origina, como recordaremos, la racemización total.]
Pues bien, si el ataque a ambas caras del carbocatión fuera puramente
aleatorio, esperaríamos obtener cantidades iguales de los dos enantiómeros, es
decir, sólo la modificación racémica. No obstante, a pesar de que a veces la
racemización es muy elevada 90% o más, casi nuncca es completa y, en general,
el producto invertido excede a su enantiómero: la reacción procede con
racemización más algo de inversión neta.
¿Cómo podemos acomodar incluso
esta inversión neta limitada dentro del marco del mecanismo SN1?
¿Cómo justificamos el hecho de que el ataque al carbocation no sea puramente
aleatorio? Es evidente que el exceso de inversión se debe, de alguna forma, al grupo
saliente: debe ejercer aún cierto control sobre la estereoquímica. En ausencia
completa del grupo saliente, el carbocatión plano perdería toda su quiralidad y
no podría generar un producto con ninguna actividad óptica. (Recuérdese: la síntesis de compuestos
quirales a partir de reactivos aquirales siempre de la modificación racémica.)
¿Cómo está involucrado el grupo saliente? Para hallar una respuesta a
esto, consideremos el proceso de la heterólisis. A medida que la reacción
avanza, la distancia entre carbono y halógeno aumenta progresivamente, hasta
que, finalmente, el enlace covalente se rompe. Se generan los dos iones de
carga opuesta pero no, de inmediato, en forma de iones completamente libres. Inicialmente deben encontarse
muy juntos, lo suficiente para que la atracción electrostática sea apreciable;
existen así por algún tiempo como un par
iónico (Sec. 6.4). Recién formados, los iones se encuentran en contacto
mutuo. Luego, a medida que se alejan por difusión, comienza a intervenir una
capa tras otra de disolvente, hasta que por fin son mutuamente independientes y
se habla de iones <<libres>>.
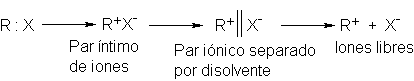
Ahora, el ataque nucleofílico puede suceder en cualquier momento
después de la heterólisis, por lo que puede involucrar a cualquier especie,
desde el par iónico formado inicialmente, hasta el carbocatión libre. El ataque
sobre este último es el azar, dando modificación racémica. Sin embargo, el
ataque sobre el par iónico no es el azar: el anión se encuentra más o menos
cerca del frente del carbocatión, protegiendo así este lado del ataque; el
resultado es el ataque preferencial por atrás. Por tanto, mientras ocurra el
ataque antes de la separación completa del par iónico, la inversión de la
configuración compite con la racemización.
De esta manera, el mecanismo SN1 puede justificar el hecho
de que la racemización no es completa. Pero lo principal es contraste
importante con la estereoquímica SN2 que la racemización suceda
después de todo. A diferencia de una reacción SN2 que procede con
inversión completa, una reacción SN1
lo hace con racemización.
Que haya dos clases de estereoquímica, refuerza la idea central de que
existen dos mecanismos diferentes. El aspecto particular de la estereoquímica
apoya poderosamente los mecanismos específicos mencionados. La inversión
completa en la reacción SN2 afirma la idea de la formación y ruptura
concertada de enlaces, en una sola etapa;
la racemización en la reacción SN1 indica que la formación y ruptura
de enlaces suceden por separado, en etapas
distintas.
El próximo aspecto que estudiaremos de la reacción SN1 es
el problema de la reactividad, para
cuya comprensión debemos volver otra vez a la química de los carbocationes, y
examinar lo que, para nosotros, será su propiedad más importante. Sus estabilidades relativas.
5.20 Estabilidades relativas de carbocationes
Cuando quisimos comparar estabilidades de radicales libres (Sec.
3.24), utilizamos las energías de disociación homólitica de enlaces, puesto que
éstas son las que intervienen en reacciones
con generación de radicales libres.
Queremos comparar ahora estabilidades de carbocationes, y para hacerlo
seguiremos la misma línea de razonamiento que para los radicales libres. Esta
vez, sin embargo, debemos partir de las energías de disociación heterolítica de enlaces de la tabla 1.3
(Sec. 1.14), puesto que éstas se aplican a reacciones con generación de
carbocationes. En esta tabla se registran energías para enlaces que unen bromo
con varios grupos. Estos valores son los H de las reacciones siguientes:
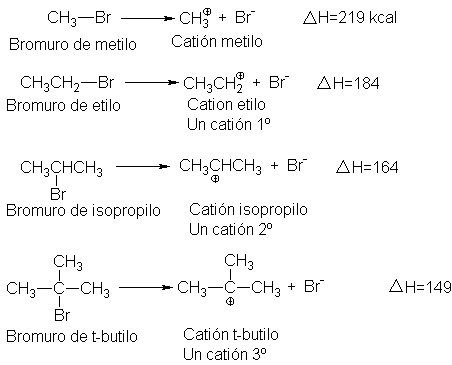
Por definición, esta energía de disociación de enlace es la cantidad
de energía que debe suministrarse para convertir un mol de bromuro de alquilo
en carbocationes y iones bromuro.
![]()
Podemos apreciar que la cantidad de energía necesaria para generar las
diversas clases de carbocationes decrece en el orden:CH3+ > 1º > 2º > 3º.
Si se necesita menor energía para formar un carbocatión que otro, sólo
puede significar que, en relación con el
bromuro de alquilo del cual se forma, un carbocatión contiene menor energía
que el otro, es decir, es más estable
(véase Fig. 5.9).
No intentamos comparar, por ejemplo, los contenidos energéticos
absolutos de los cationes isopropilo y t-butilo, simplemente sostenemos que la
diferencia en energía entre el bromuro de isopropilo y sus cationes
correspondientes es mayor que la diferencia entre el bromuro de t-butilo y sus
carbocationes. Cuando comparamos
estabilidades de carbocationes , comprendemos que nuestro estándar para
cada catión es el sustrato del cual se
deriva. Este tipo de estabilidad que nos interesa lo trataremos más
adelante.
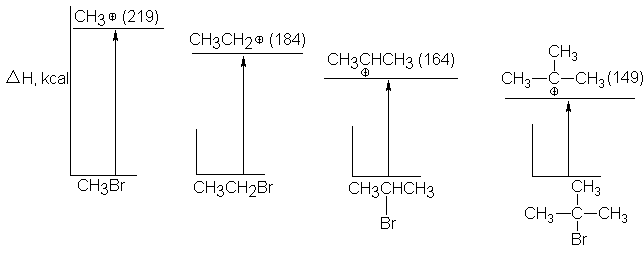
Utilizamos bromuros de alquilo para nuestra comparación, pero igualmente
pudimos emplear fluoruros, cloruros o yoduros de alquilo, o los alcoholes
correspondientes. Las energías de disociación de enlaces de la tabla 1.3
muestran para todos estos compuestos el mismo orden de estabilidad de
carbocationes. Incluso las magnitudes de diferencias energéticas, en kcal/mol,
son casi las mismas, cualquiera que sea la clase de compuestos de origen. La
diferencia entre los cationes metilo y t-butilo, por ejemplo, enr elación con
diversos sustratos, es: fluoruros, 67 kcal; cloruros, 70 kcal; bromuros, 70
kcal; yoduros, 72 kcal, y alcoholes, 66 kcal.
En relación con el sustrato del cual se forma cada uno, el orden de
estabilidad de carbocationes es:
![]()
Descubriremos que este mismo orden no sólo se aplica a los
carbocationes cuando se forman por heterólisis, sino también cuando se forman
mediante procesos enteramente diferentes.
Las diferencias de estabilidad entre carbocationes es mucho mayor que entre radicales libres.
El radical libre t-butilo, por ejemplo, es sólo 12 kcal más estable que el
metilo; el catión t-butilo es, según el sustrato, 66-72 kcal más estable que el
catión metilo. Estas diferencias, mucho mayores, en estabilidad, dan origen a
efectos mucho mayores en la reactividad.
Hasta este momento, nuestro análisis se ha basado en energías de
disociación de enlaces, las cuales se miden en la fase gaseosa. Sin embargo,
casi toda la química de carbocationes se desarrolla en solución y, como
sabemos, los disolventes pueden ejercer efectos estabilizantes poderosos en
solutos iónicos. ¿Es válido el orden de estabilidades establecido para
carbocationes en solutos iónicos.¿Es
válido el orden de estabilidades establecido para carbocationes en solución? Se
da respuesta a esto en forma más directa, midiendo, en varios disolventes, los
H para la generación de carbocationes, utilizando el método del superácido de
Olah. Los valores obtenidos revelan el mismo orden para la estabilidad de
carbocationes, en relación con el sustrato de origen, que las energías de
disociación. Incluso las diferencias en
estabilidad, en kcal/mol, son prácticamente las mismas.
![]()
Hemos logrado así un orden de estabilidades de carbocationes que vale
tanto para soluciones como para la fase gaseosa, y que es aplicable a la
generación de carbocationes con una amplia variedad de sustratos y en una vasta
gama de reacciones químicas. A medida que avancemos nuestro estudio, añadiremos
otros tipos de carbocationes a nuestra serie y examinaremos otros tipos de
reacciones mediante las que pueden ser generados.
Veamos ahora cómo puede justificarse este orden.
5.21 Estabilización de carbocationes. Acomodación de
la carga.
Efectos polares
El rasgo característico de un carbocatión es, por definición, su
carbono deficiente en electrones y su correspondiente carga positiva. La
estabilidad relativa de un carbocatión está determinada principalmente por su
mayor o menor capacidad para acomodar
dicha carga.
De acuerdo con las leyes de la electrostática, la estabilidad de un sistema cargado aumenta con la dispersión de la
carga. En consecuencia, todo factor que tiende a esparcir la carga positiva
del carbono deficiente en electrones, y distribuirla sobre el resto del ión,
debe estabilizar al carbocatión.
Consideremos un sustituyente, G, ligado a un carbono electrónicamente
deficiente, en lugar de un átomo de hidrógeno. En comparación con este último,
G puede liberar, o aceptar, electrones. Tal
efecto sobre la disponibilidad de electrones en el centro reactivo se denomina efecto polar.

Un sustituyente que libera electrones
tiende a reducir la carga positiva en el carbono deficiente en
electrones; al hacer esto, el propio sustituyente se torna algo positivo: la dispersión de la carga
estabiliza al carbocatión.
Un sustituyente que atrae electrones tiende a intensificar la carga
positiva en el carbono deficiente en electrones, así el carbocatión resulta
menos estable.Acabamos de ver que el orden de estabilidad de carbocationes es
el siguiente:
![]()
Por definición, la distinción entre cationes primero, secundario y
terciario es el número de grupos alquilo ligados al carbono con deficiencia
electrónica. Luego, los hechos establecen
que, cuanto mayor es el número de
grupos alquilo, más estable es el carbocatión.
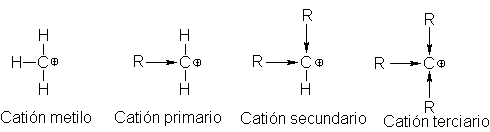
Si nuestra generalización sobre dispersión de carga tiene aplicación
en este caso, quiere decir que los grupos alquilo liberan electrones.
¿Es la liberación de electrones lo que hubiéramos esperado de grupos
alquilo? Ingold (Sec. 5.12) ha propuesto que los grupos alquilo, al carecer de
tendencia polares fuertes propias, pueden realizar prácticamente todo lo que
otros grupos presentes en la molécula les demande. La acumulación de hechos
prueba que efectivamente es así: a menudo, los grupos alquilo tienden a
estabilizar tanto a cationes, como a aniones, indicando liberación o atracción
de electrones según sea la demanda.
En un carbocatión, el carbono con deficiencia electrónica necesita urgentemente
electrones es como si fuera un elemento distinto muy electronegativo, por lo
que induce a los grupos alquilo a
liberarlos para satisfacer esa necesidad.
Ahora,¿cómo ejerce un sustituyente su efecto polar? A pesar de la
cantidad ingente de trabajo realizado y que sigue realizando , no hay acuerdo
general, excepto en que deben operar por lo menos dos factores. Consideremos
que la atracción o liberación de electrones es el resultado de la operación de
dos factores: el efecto inductivo y el de
resonancia:
El efecto inductivo depende de la tendencia <<intrínseca>> de un sustituyente
a liberar o atraer electrones por definición, su electronegatividad, y actúa a
lo largo de la cadena molecular o a través del espacio. El efecto se debilita
gradualmente con el aumento de la distancia al sustituyente. La mayoría de los elementos que suelen sustituir
hidrógeno en una molécula orgánica son más electronegativos que el hidrógeno,
de modo que estos sustituyentes suelen
ejercer efectos inductivos de atracción de electrones: por ejemplo: -F, -CI,
-Br, -I, -OH, -NH2, -NO2.
El efecto de resonancia
implica deslocalización de electrones
típicamente los denominados electrones (pi)-. Depende del solapamiento de
ciertos orbitales, por lo que sólo puede operar cuando el sustituyente se
encuentra localizado de ciertos modos especiales en relación con el centro de carga.
Por su naturaleza, el efecto de resonancia es estabilizante (Sec. 10.14), de
modo que supone una extracción de electrones de un centro cargado negativamente
y a una liberación de electrones hacia un centro con carga positiva.
La naturaleza de la liberación de electrones por los grupos alquilo no
está clara. Puede que se trate de un efecto inductivo; puede ser uno de
resonancia (hiperconjugación, Sección
10.16), siendo proporcionados los electrones por solapamiento de enlaces o con
el orbital p vacío de un carbono deficiente en electrones. Puede muy bien ser
una combinación de ambos. Cuando nos referimos en este texto al efecto
inductivo de grupos alquilo, debe entenderse que éste puede incluir una
contribución de la hiperconjugación.
Como quiera que se genere, el efecto polar de los grupos alquilo no es
poderoso, sin embargo da como resultado diferencias muy grandes de estabilidad
entre las diversas clases de carbocationes. Estas diferencias debemos tenerlas
muy presentes al ocuparnos de la química de los carbocationes, que es muy
variada.
5.22 Reacción SN1:
reactividad.
Facilidad de formación de
carbocationes
Volvamos una vez más a la sustitución nucleofílica y a la pregunta de
cómo la estructura del grupo alquilo afecta a la reactividad. Ya vimos (Sec.
5.15) que la reactividad SN2 disminuye en la serie CH3W,
1º, 2º, 3º, como postularon Hughes e Ingold (Sec. 5.12). Veamos ahora cuáles
son los hechos en relación con la otra mitad de su teoría dual: ¿cambia la
reactividad por SN1 en el sentido contrario a lo largo de la misma
serie?
En condiciones que favorezcan ampliamente la SN1, se
obtienen los resultados siguientes:
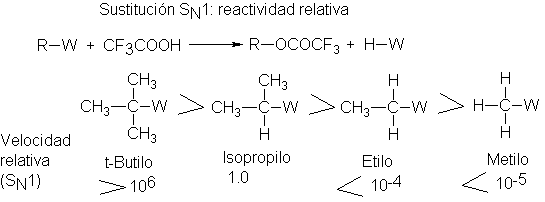
Se confirma así el orden de reactividades postulado. También, las diferencias
en reactividad son, como se señaló, mucho más grandes que las que se
encontraron para la reacción SN2 véase la curva SN1
fuertemente ascendente de la figura 5.1 (Sec. 5.12). Pos SN1, los
sustratos terciarios son más de un millón de veces más reactivos que los
secundarios, que a su vez son por lo menos 10 000 veces y es probable que más
de un millón más reactivos que los primarios.
Incluso se cree que estas diferencias están subestimadas. Las
reactividades de los sustratos primarios y metílicos son muy inferiores a los
valores máximos indicados; es probable que aun las bajas velocidades medidas
para ellos son en gran parte SN2, no para SN1, actuando
el disolvente como nucleófilo (Sec. 6.9).
![]() En consecuencia, la reactividad de los sustratos en la reacción SN1
sigue la secuencia:
En consecuencia, la reactividad de los sustratos en la reacción SN1
sigue la secuencia:
Ahora bien, el paso determinante de la velocidad en SN1 es la formación del
carbocatión; esto es, un sustrato sufre SN1 más rápido que otro
porque forma el carbocatión más rápido.
Por tanto, nuestra secuencia de reactividad conduce directamente a otra que indica la velocidad relativa de formación de carbocationes:
![]()
Al ordenar los carbocationes de acuerdo con su velocidad de formación,
lo hemos hecho simultáneamente en el orden de sus estabilidades. Cuanto más estable es un carbocatión, más
rápida es su formación.
Probablemente ésta es la generalización más conveniente de la
estructura y reactividad que aparece en este libro o que de hecho existe en la
química orgánica. Los carbocationes se generan a partir de muchos compuestos
distintos de los halogenuros de alquilo, y en reacciones diferentes de la
sustitución nucleofílica. No obstante, en
todas estas reacciones, en las que se generan carbocationes, la estabilidad de
éstos desempeña un papel importante en el
gobierno de la reactividad y la orientación.
¿Cómo podemos demostrar el hecho de que la velocidad de formación de
un carbocatión depende de su estabilidad? Como siempre, para dar respuesta a
esto, debemos considerar la reacción específica implicada en este caso, la SN1
y comparar la estructura de los reactivos con la del estado de transición.
En una reacción SN1 de un halogenuro de alquilo, el
carbocatión se forma por heterólisis de la molécula del sustrato; esto es, por
ruptura del enlace carbono-halógeno. En el reactivo, el carbono y el halógeno
comparten un par de electrones; salvo por una polaridad moderada, estos dos
átomos son neutros. En los productos, el halógeno se ha llevado consigo el par
de electrones, quedando el carbono con sólo un sexteto: el halógeno es portador
de una carga negativa completa, mientras que el carbocatión tiene una carga
positiva neta centrada en un carbono.
En el estado de transición, el enlace C-X debe estar parcialmente
roto, y el halógeno ya ha quitado parcialmente el par de electrones al carbono.
Aquél ha ganado parcialmente la carga negativa que ha de llevar en el ión
halogenuro. Es más, el carbono ha ganado
parcialmente la carga positiva que ha de llevar en el carbocatión.
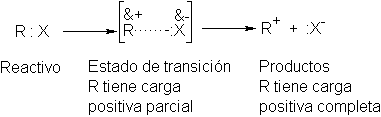
Los grupos liberadores de electrones tienden a dispersar la carga
positiva parcial (&+) que se está desarrollando sobre el
carbono, estabilizando así el estado de transición. La estabilización de éste
rebaja la Eact y permite una reacción más rápida (véase Fig. 5.10).
De esta manera, y según el grado de ruptura del enlace C-X, el grupo
alquilo posee el carácter del carbocatión que ha de originar. El mismo factor,
la liberación de electrones, que estabiliza al carbocatión, también estabiliza
al carbocatión incipiente en el
estado de transición.
En 1979, Edward Arnett (Universidad de Duke) y Paul Schleyer
(Universidad de Erlangen-Nurnberg)
publicaron <<una corroboración extraordinaria de la validez fundamental
de la “teoría carbocatiónica de la química orgánica”>>. Para un conjunto
de sustratos de estructuras ampliamente diferentes, compararon los valores de
las Eact de reacciones SN1 con los calores de ionización
en soluciones de superácidos, y hallaron una dependencia cuantitativa directa entre la velocidad de formación de
carbocationes y la estabilidad de éstos.
Confirmaron que el carbocatión más estable se forma más velozmente.
A medida que nos enfrentamos a otras reacciones con formación de
carbocationes, debemos examinar para cada una de ellas la estructura del estado
de transición. En la mayoría de estas reacciones, sino en todas, descubriremos
que el estado se distingue de los reactivos principalmente por parecer al
producto. El carácter carbocatiónico del
estado de transición será el factor que más afecta a la Eact; por
consiguiente, cuanto más estable es el carbocatión, más estable será el estado
de transición que conduce aquel, y más rápido se formará el carbocatión.
Sin embargo, lo estudiado aquí será aplicado de un modo aún más
general. Retornaremos una y otra vez a la relación entre efectos polares y
dispersión de carga, y entre esta última y estabilidad. Hallaremos que estas
relaciones no sólo nos ayudarán a comprender reacciones carbocatiónicas de muchos tipos, sino también todas aquellas
en las que se desarrolla o desaparece una carga bien positiva o negativa. Estas
incluirán reacciones aparentemente tan distintas de la sustitución SN1
como la deshidratación de alcoholes, la adición a alquenos y la sustitución
aromática tanto electrofílica como nucleofílica y las propiedades fundamentales
de acidez y basicidad.
Por consiguiente, las diferencias en la reactividad por SN1
dependen de las diferencias de
estabilidad entre las diversas clases de carbocationes. En la siguiente sección
veremos que estas diferencias de estabilidad llevan a un comportamiento
sorprendente de los carbocationes.
5.23
Transposición de carbocationes
Anteriormente dijimos que el desarrollo de la teoría de los
carbocationes se debe a un esquema de comportamiento. La característica más
destacada de ese esquema es la ocurrencia de transposiciones.
Por ejemplo, en la sustitución nucleofílica se observa a veces que el
grupo entrante, Z, se une a un carbono distinto al que originalmente estaba
ligado el grupo saliente, X. Por ejemplo:
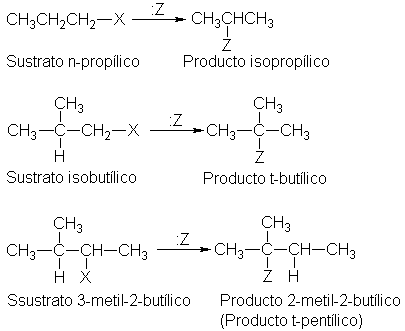 Ejemplo:
Ejemplo:
En cada uno de estos casos podemos observar que, para acomodar Z en la
posición nueva, debe producirse una transposición de átomos de hidrógeno en el sustrato.
La transformación de un grupo n-propilo a uno isopropílico, por ejemplo,
requiere la eliminación de un H del C-2, y la unión de un H al C-1.
Aveces, se produce incluso una transposición del esqueleto carbonado.
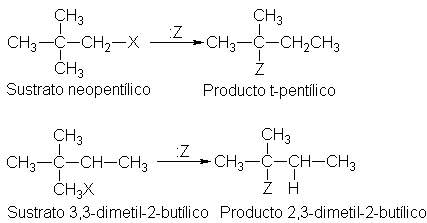
En reacciones de tipos diferentes eliminación, adición, también se
observan transposiciones similares a las presentadas. Esta similitud de
comportamiento plantea una similitud en el mecanismo. Por muy distintos que
sean los diversos mecanismos, todos tienen una característica común: en alguna
etapa se forma el mismo intermediario, y es éste el que sufre la transposición.
Este intermediario, propuesto claramente por primera vez en 1922 por Hans
Meerwein (Sec. 5.17), es el carbocatión.
Pues bien, de los dos mecanismos propuestos para la sustitución
nucleofílica, solamente el SN1 se postula que involucra un
carbocatión intermediario, por lo que solo esperamos que las reacicones
procedentes de SN1 se acompañen por estas transposiciones
características. En contraste, el paso único postulado para SN2,
sencillamente no proporciona oportunidades para tales transposiciones.
La suposición anterior se corrobora experimentalmente, como se ilustra
en el siguiente ejemplo. Los sustratos de neopentilo son particularmente propensos
a transponerse a productos t-pentílicos: en una solución de etóxido de sodio en
etanol, el bromuro de neopentilo sufre una reacción (lenta) de segundo orden, generado éter etil
neopentil, no transpuesto; en una solución que sólo contiene etanol, sufre una
reacción (lenta) con cinética de primer orden, para dar éter etil t-pentil (y
otros productos transpuestos).
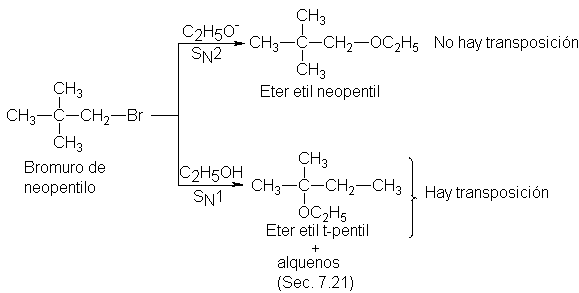
La ocurrencia y ausencia de una transposición es una diferencia
notable, y proporciona otra pieza de evidencia de que hay dos mecanismos para la sustitución nucleofílica. Además, la
transposición es un apoyo poderoso para la forma particular del mecanismo SN1
la mediación de carbocationes, porque liga este mecanismo a otros de aquellas
clases de reacciones en las que también se observan transposiciones. La
correlación entre transposición y cationes intermediarios es tan fuerte que, en
ausencia de otra información acerca de un ejemplo particular de sustitución
nucleofílica, se considera una transposición como evidencia de que la reacción
es SN1.
Sobre esta base, entonces, podemos explicar los productos observados
de la manera siguiente. Un sustrato n-propílico, por ejemplo, da el catión
n-propilo, el cual se transpone el catión isopropilo, que se combina con el
nucleófilo para dar el producto de isopropilo.
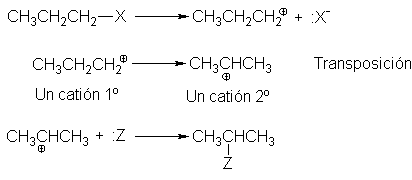
De manera similar, el catión isobutilo se transpone al t-butilo,
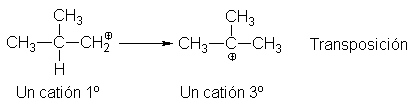
El catión 3-metil-2-butil se transpone al 2-metil-2-butilo,
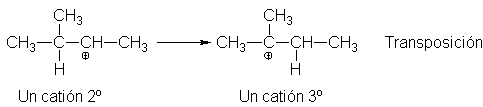
El catión neopentilo se transpone al t-pentilo,
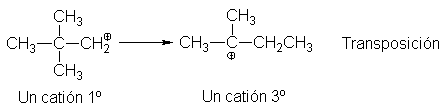
Y el catión 3,3-dimetil-2-butilo se transpone al 2,3-dimetil-2-butilo.
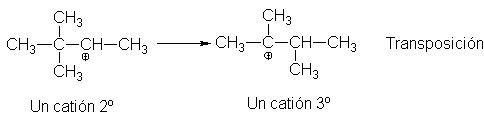
Podemos observar en cada caso,
que la transposición se realiza de manera tal que un catión menos estable se
convierte en otro más estable: uno primario en uno secundario, uno primario
en uno terciario, o uno secundario en uno terciario.
¿Cómo sucede esta transposición? Frank Whitmore (Universidad del
Estado de Pensilvania)describió la transposición así: un átomo de hidrógeno o
un grupo alquilo migra con un par de
electrones desde un carbono que pierde al grupo migratorio adquiere la
carga positiva. Una migración de hidrógeno con un par de electrones se conoce
como desplazamiento de hidruro; una
migración similar de un grupo alquilo es un
desplazamiento de alquilo. Estos son sólo dos ejemplos del tipo más común
de transposición, los desplazamientos
1,2: las transposiciones en las que el grupo que migra se desplaza de un
átomo al vecino inmediatamente.
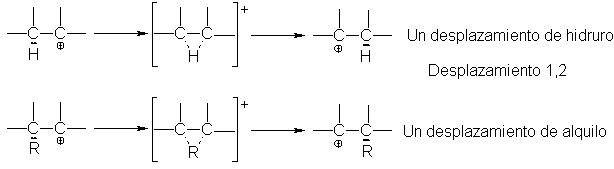
Podemos explicar las transposiciones en reacciones SN1 como
sigue. Se forma un carbocatión por pérdida del grupo saliente del sustrato. Si un desplazamiento 1,2 de hidrógeno o
alquilo puede generar un carbocatión más estable, entonces tiene lugar esa
transposición. El carbocatión nuevo se combina ahora con el nucleófilo,
para dar el producto de la sustitución.
En el caso del catión n-propilo, por ejemplo, un desplazamiento de
hidrógeno da el más estable catión isopropilo; la migración de un metilo sólo
formaría un catión n-propilo diferente.
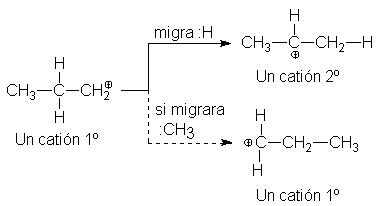
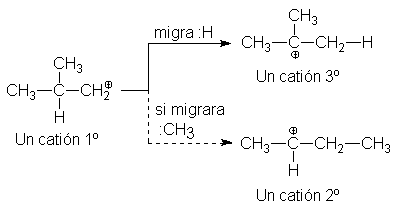 En el catión isobutilo, el desplazamiento de hidruro da un catión
terciario, por lo que se prefiere a una migración de metilo, que sólo generaría
un catión secundario.
En el catión isobutilo, el desplazamiento de hidruro da un catión
terciario, por lo que se prefiere a una migración de metilo, que sólo generaría
un catión secundario.
En el caso del catión 3,3-dimetil-2-butilo, por otra parte, un
desplazamiento de metilo puede dar un catión terciario, y es la transposición
que tiene lugar.
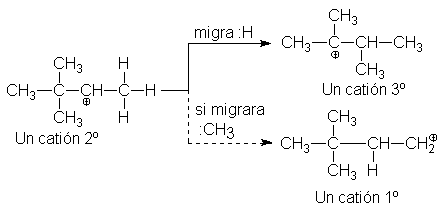
Podemos imaginar la transposición como una reacción ácido-base
intramolecular, en la que el ácido más fuertecomo es usual, toma la base. La
base es el grupo migratorio con sus electrones (hidruro o alquilo). Compiten
por ella dos ácidos de Lewis: lso carbonos electrónicamente deficientes en los
carbocationes alternativos. En la transposición n-isopropilo, por ejemplo, es
más deficiente en electrones el C-1 y, por consiguiente, es el ácido más
fuerte, por lo que termina tomando la base.
Al igual que se comprobó la realidad de los carbocationes, también se
comprobó la de su transposición. Preparados en condiciones superácidas del
método de Olah, y estudiados mediante la espectroscopia, ha podido observarse la transposición de carbocationes; ha podido
medirse la velocidad de algunas transposiciones y se han calculado sus Eact.
Si se añade agua, ésta se combina con el catión transpuesto, apareciendo el OH
en una nueva posición en la molécula. Nuevamente observamos aquí los pasos
propuestos para SN1 como etapas individuales, pero esta vez con
transposición: primero, la formación de un carbocatión; después, su
transposición a un nuevo catión, y, finalmente, la combinación de este catión
transpuesto con el nucleófilo.
En nuestra breve familiaridad con el carbocatión, hemos conocido dos
de sus reacciones. Un carbocatión puede:
(a) combinarse con un nucleófilo;
(b) transponerse a un carbocatión más estable.
Esta lista crecerá rápidamente.
En una transposición, como en todas las demás reacciones de un
carbocatión, el átomo de carbono deficiente en electrones gana un par de éstos,
a expensas ahora de un átomo de carbono vecino, uno capaz de acomodar mejor la
carga positiva.
En el capítulo 6 veremos otro aspecto de la reacción SN1 y
examinaremos el factor que hace posible todo: el disolvente. Por ahora,
regresemos al punto inicial, la competencia entre SN1 y SN2.
Hasta el momento hemos descrito dos mecanismos para la sustitución
nucleófilica: el SN2, caracterizado por
(a) una cinética de segundo orden,
(b) inversión estereoquímica completa,
(c) ausencia de transposiciones, y
(d) la secuencia de reactividades CH3W >1º > 2º > 3º;
y el SN1, caracterizado por
(a) una cinética de primer orden,
(b) racemización,
(c) transposiciones, y
(d) la secuencia de reactividades 3º> 2º> 1º > CH3W.
Excepto la mención breve de la sección 5.12, hemos tratado estos
mecanismos como temas separados. Estudiemos ahora la relación entre ambos. Para
un sustrato dado y un determinado conjunto de condiciones, ¿cuál será el
emcanismo que seguirá una reacción?, ¿qué podemos hacer, en todo caso, para
dirigirla hacia uno u otro mecanismo?
Para responder a estas preguntas, consideremos lo que sucede a una
molécula del sustrato. Puede sufrir un ataque por atrás por el nucleófilo, o
bien someterse a una heterólisis, con formación de un carbocatión. El proceso
que sea más rápido será el que determine el mecanismo predominante. (Recuérdese: heterólisis es el primer
paso del mecanismo SN1 y determinante de la velocidad.) Una vez más,
debemos prestar atención a la velocidad
relativa de las reacciones que compiten.
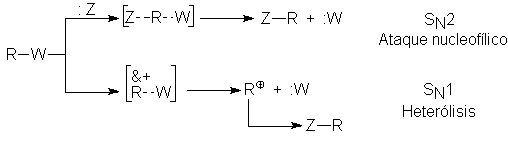
Examinemos cada uno de los componentes del sistema de reacción y veamos
el efecto que ejerce sobre esta competencia entre el ataque nucleofílico y la
heterólisis.
Comencemos con el sustrato,
que consta de dos partes: el grupo alquilo y el grupo saliente. La naturaleza del grupo saliente es, por
cierto, vital para que ocurra sustitución.
Cualquiera que sea el proceso que se está realizando, ataque
nucleofílico o heterólisis, se está rompiendo la unión con el grupo saliente;
cuanto más fácil resulte esta ruptura esto es, cuanto mejor sea el grupo
saliente, mas rápidamente procede la reacción. Un grupo saliente mejor acelera,
por tanto, la reacción de ambos mecanismos, y, efectivamente, acelera ambos
aproximadamente en igual magnitud. El resultado es que la naturaleza del grupo
saliente tiene poco efecto sobre el predominio de uno u otro mecanismo.
En contraposición, la naturaleza
del grupo alquilo, R, del sustrato ejerce un efecto profundo en el
mecanismo que se seguirá. En R operan dos factores: el impedimento estérico, que
determina en gran medida la facilidad del ataque por atrás; y la capacidad
para acomodar una carga positiva,
que determina preponderantemente la facilidad de la heterólisis. Procediendo en
las serie de alquilos simples CH3, 1º, 2º, 3º, el grupo R resulta
por definición, más ramificado. Hay un aumento regular en el número de
sustituyentes sobre el carbono: sustituyentes voluminosos, liberadores de
electrones. Aumenta el impedimento estérico, con lo que el ataque por atrás se
hace más difícil y, por consiguiente, más lento. Al mismo tiempo aumenta la
capacidad para acomodar una carga positiva: la heterólisis se hace más fácil y
rápida.

El resultado es el esquema que ya hemos visto: para sustratos
metílicos y primarios, una predisposición hacia SN2; para
terciarios, una predisposición hacia SN1. Para los sustratos
secundarios, hay una tendencia hacia un comportamiento intermediario: una
mezcla de ambos mecanismos o, como veremos en la sección siguiente, quizás un
mecanismo con características tanto de SN2 como de SN1.
A pesar de esta predisposición de un sustrato en particular hacia uno
u otro mecanismo, podemos controlar el curso de una reacción en un grado
considerable mediante nuestra selección de condiciones experimentales. Para
apreciar cómo puede lograrse esto, examinaremos los demás componentes del
sistema reaccionante.
Analicemos ahora el nucleófilo.
La diferencia clave entre los mecanismos SN2 y SN1 reside
realmente en cuándo participa el nucleófilo: en el paso que determina la
velocidad del SN2, pero después del paso determinante de la
velocidad de SN1. Esta
diferencia en la secuencia conduce directamente a dos factores que contribuyen
a precisar el mecanismo que ha de seguirse: la concentración del nucleófilo y su naturaleza.
La velocidad de SN2 depende de la concentración del
nucleófilo, [:Z] ; sabemos que la reacción es de segundo orden (Sec. 5.13).
![]()
La velocidad de SN1 es independiente de [:Z] ; la reacción
es de primer orden (Sec. 5.16).
![]()
Un aumento en [:Z] acelera la reacción de segundo orden, pero no tiene
ningún efecto sobre la de primer orden: la fracción de la reacción por SN2
aumenta. Una disminución de [:Z] retarda la reacción de segundo orden, pero no
ejerce efecto sobre la de primer orden: disminuye la fracción de la reacción
por SN2. El resultado neto es que, a igualdad de otras condiciones, una concentración elevada del nucleófilo
favorece la reacción SN2, y una concentración baja favorece la
reacción SN1.
La velocidad de SN2 depende, igualmente, de la naturaleza
del nucleófilo: uno más fuerte ataca más velozmente al sustrato. La velocidad
de SN1 es independiente de la naturaleza del nucleófilo: más fuerte o más débil, el
nucleófilo espera hasta que se haya formado el carbocatión. El resultado neto
es que, a igualdad de otras cosas, un nucleófilo fuerte favorece la reacción SN2,
y un nucleófilo débil favorece la reacción SN1.
Ya tuvimos ocasión de apreciar
este efecto en la sección 5.23. El bromuro de neopentilo reacciona con el
nucleófilo fuerte, etóxido, para generar neopentil etil éter no transpuesto, lo
que claramente es una reacción SN2. Reacciona con el nucleófilo
débil, etanol, para dar t-pentil etil éter transpuesto: es, sin lugar a dudas,
una reacción SN1.
No hemos considerado el tercer componente del sistema, el que ofrece
el mayor campo de acción para el control de una reacción: el disolvente. En el capítulo 6
examinaremos detalladamente su papel.
Al comenzar este capítulo dijimos que una reacción química es el
resultado de una competencia: lo que
realmente sucede cuando se mezcla un conjunto de reactivos en ciertas
condiciones particulares es aquello que ocurre más rápido. Hasta ahora nos ha interesado la competencia que hay
entre dos caminos diferentes para la sustitución nucleofílica. En el capítulo 7
encontramos una competencia adicional: entre la sustitución nucleofílica y un
tipo de reacción totalmente diferente: la eliminación. En todo esto, lo que nos
interesa son los factores que favorecen un mecanismo o un tipo de reacción
sobre otra y Un, cuando es posible, lo que podemos hacer para controlar el
resultado.
5.25 Reacción de alcoholes
con halogenuros de hidrogeno.
Catálisis ácida
Un método para obtener halogenuros de alquilo se da empleando la reacción
de alcoholes con halogenuros de hidrógeno (Sec. 5.7). Estudiemos con más
detenimiento esta reacción, no sólo como método de síntesis importante, sino
como ejemplo de una sustitución nucleofílica.
Al hacer esto, veremos algo que resulta completamente nuevo: la manera
de transformar instantáneamente, un
grupo saliente pobre en otro muy bueno, y sin más esfuerzo que el de verte una
solución de una botella en un matraz. Veremos el tipo más importante y más
simple de efecto catalítico que conoce el químico orgánico: uno que tiene un
papel clave en la química de compuestos de todo tipo, tanto en el tubo de ensayos, como en el organismo vivo.
Los alcoholes reaccionan con facilidad con los halogenuros de
hidrógeno para dar halogenuros de alquilo y agua. La reacción se produce
haciendo pasar halogenuro seco por el alcohol o calentado el alcohol con el ácido acuoso concentrado. En
presencia del alcohol, a veces se genera bromuro de hidrógeno mediante la
reacción entre ácido sulfúrico y bromuro de sodio.
El menos reactivo de los halogenuros de hidrógeno, el HCI, requiere,
por lo general, la presencia de cloruro de cinc para reaccionar con alcoholes
primarios y secundarios; por otra parte, el alcohol t-butílico, muy reactivo,
se convierte en el cloruro por simple agitación con ácido clorhídrico
concentrado a temperatura ambiente. Ejemplos:
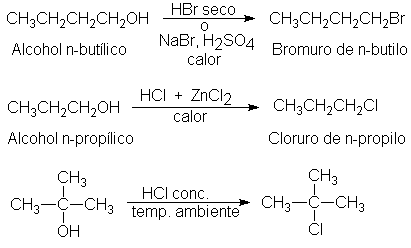
Enumeremos algunos hechos conocidos acerca de la reacción entre alcoholes y halogenuros de hidrógeno.
(a) La reacción es catalizada
por ácidos. A pesar de que los propios halogenuros de
hidrógeno acuoso son ácidos fuertes, la presencia de ácido adicional acelera la
formación de halogenuros.
(b) Hay transposición del
grupo alquilo, salvo en la mayoría de los alcoholes primarios.
El grupo alquilo del
halogenuro no tiene siempre la misma estructura que en el alcohol original. Por
ejemplo:
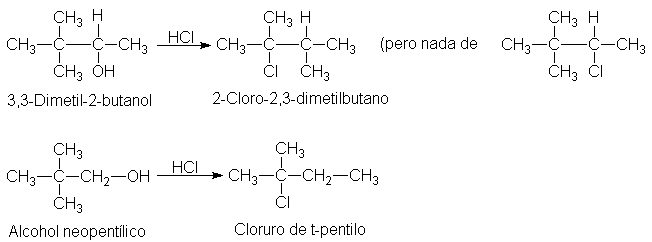
Podemos apreciar que el halógeno no siempre se une al carbono que originalmente
tenía el hidroxilo (primer ejemplo); aún puede ser diferente el esqueleto
carbonato del producto al del material de partida (segundo ejemplo).
Por otra parte, y tal como se ilustró para los alcoholes n-propílico y
n-butílico, la mayoría de los alcoholes primarios dan altos rendimientos en
halogenuros primarios sin
transposición.
(c) El orden de reactividad de
los alcoholes con HX es 3º > 2º > 1º < CH3. La reactividad decrece a lo largo de la mayor parte de la serie
(este orden es la base del ensayo de
Lucas, Sec. 18.9), pasa por un mínimo
en 1º y aumenta de nuevo en CH3.
¿Qué nos sugieren los hechos recién enumerados en cuanto al mecanismo
de la reacción entre alcoholes y halogenuros de hidrógeno?
La catálisis por ácido
sugiere que está involucrado el alcohol protonado ROH2+.
Las transposiciones establecen que los carbonationes son intermedios aunque no
con alcoholes primarios. La idea de los carbocationes se apoya en el orden de reactividad de los alcoholes, que
corre paralelo a la estabilidad de los carbocationes, excepto para el metilo.
Basándonos en estas evidencias, formulamos el mecanismo siguiente:
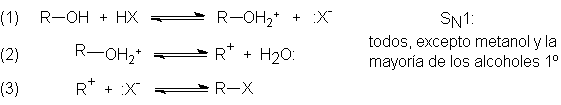
El alcohol acepta el ión hidrógeno para formar la especie protonada
(paso 1), que se disocia en agua y un carbocatión (paso 2); el carbocatión se
combina entonces con un ión halogenuro (no necesariamente con el del paso 1)
para formar el halogenuro de alquilo (paso 3).
Observando el mecanismo expuesto, reconocemos la naturaleza de la
reacción: sustitución nucleofílica,
con el alcohol protonado como sustrato y el ión halogenuro como nucleófilo. Una
vez reconocido el tipo de reacción, las evidencias de la información encajan.
El conjunto de ecuaciones expuesto arriba es, por supuesto, el mecanismo
para la sustitución SN1. Los alcoholes primarios no se transponen,
simplemente porque no reaccionan por este mecanismo, sino por el SN2:
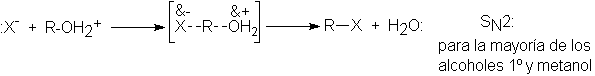
Tenemos aquí otro ejemplo de la característica de la sustitución
nucleofílica: un cambio en la molecularidad de la reacción, en este caso
particular entre 2º y 1º. Se confirma este cambio porque la reactividad pasa
por un mínimo en 1º y aumenta nuevamente en el metilo.
Comenzando con el extremo metilo de la serie, revisemos lo que
probablemente suceda aquí. El sustrato metílico es el menos capaz de
heterólisis y el más abierto para el ataque nucleofílico: reacciona por un
mecanismo SN2 total. También lo hacen los sustratos primarios, pero,
debido a un mayor impedimento estérico, reaccionan más lentamente que el metilo.
Los sustratos secundarios presentan un impedimento estérico aún mayor, pero
tienen mayor capacidad para generar carbocationes. La heterólisis es más rápida
para ellos que un ataque nucleofílico por un ión halogenuro, por lo que el
mecanismo cambia aquí a SN1. Con el cambio de mecanismo comienza a aumentar la velocidad.
También los sustratos terciarios reaccionan por un mecanismo SN1;
reaccionan con mayor velocidad que los sustratos secundarios, debido a la mayor
dispersión de carga en los carbocationes incipientes.
Hasta ahora hemos tratado esta reacción en función de la
clasificación, muy útil, de los sustratos en primarios, secundarios o
terciarios. No obstante, debemos tener presente que esta clasificación como tal
no es lo importante, sino los factores que realmente operan; en esta reacción,
el impedimento estérico al ataque nucleofílico y la dispersión de carga en el carbocation incipiente. Estos factores
dan origen entre otras cosas a la
relación entre 1º, 2º, 3º, y a la
competencia SN2 SN1. Pero hacen más que esto, pueden
hacer que un sustrato de una clase actúe como uno de otra; sin embargo, tal
comportamiento es comprensible con sólo examinar las estructuras implicadas.
Veamos dos de tales ejemplos.
Como se indicó anteriormente, el alcohol neopentílico reacciona con
transposición casi completa, lo que demuestra, a pesar de ser primario, que
sigue un mecanismo carbocatiónico. Esto es contrario a nuestra generalización,
pero fácilmente explicable. Aunque el neopentilo es un grupo primario, es muy
voluminoso, de modo que los sustratos
neopentílicos sufren reacicones SN2 con mucha lentitud (Sec.
5.15). La formación del catión neopentilo es también muy lenta; sin embargo, es
mucho más rápida que la reacción bimolecular alternativa.
Nuestro segundo ejemplo involucra al 1-cloro-2-propanol. Aunque
técnicamente es un alcohol secundario, reacciona con los halogenuros de
hidrógeno de forma anormalmente lenta,
con una velocidad similar a la de un alcohol primario. Esta vez no estamos
tratando con un efecto estérico, sino con uno polar. La velocidad de una
reacción SN1 depende de la
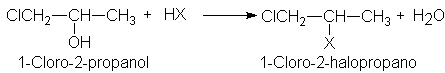
Estabilidad del carbocation que está formando (Sec. 5.22). Comparemos
el catión 1-cloro-2-propilo con uno secundario simple, por ejemplo, el catión
isopropilo. El cloro electronegativo ejerce un efecto inductivo de atracción
electrónica. Como sabemos ya (Sección 5.21), esto intensifica la carga positiva
sobre el carbono deficiente en electrones, por lo que
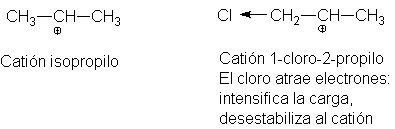
El carbocatión es menos estable. Esta misma atracción de electrones
desestabiliza al catión incipiente en el estado de transición, eleva la Eact
y retarda la reacción.
Estudiemos ahora lo que para nosotros es el aspecto más importante de
la reacción entre un alcohol y los halogenuros de hidrógeno: la catálisis
ácida. ¿Qué hace el ácido? En un primer paso, convierte al alcohol en la
especie protonada, que de hecho es el sustrato
sometido a sustitución. En ausencia de ácido, la sustitución debería
implicar por cualquiera de los dos mecanismos la pérdida de un ión hidróxido,
que es fuertemente básico y un saliente muy pobre. La sustitución con el
alcohol protonado como sustrato implica, por otra parte, la pérdida de agua que
es sólo débilmente básica y un grupo saliente muy bueno. La
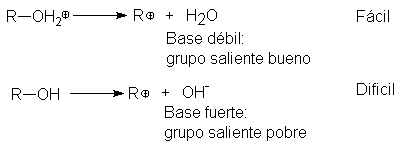
Protonación del alcohol es un simple equilibrio ácido-base, y tiene
lugar instantáneamente al mezclar los reactivos; sin embargo, transforma un
grupo saliente muy malo en uno excelente, permitiendo así que la reacción
suceda. Las pruebas indican que casi nunca ocurre la separación de un ión
hidróxido de un alcohol; las reacciones que involucran la ruptura del enlace
C-O de un alcohol en casi todos los casos parecen requerir un catalizador
ácido, cuyo propósito, como aquí, es la formación de la especie protonada.
Así, los alcoholes sufren sustitución nucleofílica, igual que los
halogenuros de alquilo, tanto por el mecanismo SN2 como por el SN1,
pero los primeros tienden más al unimolecular.
De una forma general, se puede apreciar por qué sucede esto. Para
sufrir sustitución, un alcohol debe estar protonado, lo que necesita un medio
ácido. Hemos visto que una reacción SN2 se ve favorecida por el uso
de un nucleófilo potente, algo que es muy factible en reacciones con halogenuros
de alquilo, pero no podemos tener un nucleófilo fuerte una base fuerte en el medio ácido necesario para la protonación de un
alcohol: cualquier base mucho más fuerte que el propio alcohol sería protonada
a expensas de éste. Por tanto, confinado a reaccionar con reactivos débilmente
básicos y débilmente nucleofílicos, los alcoholes reaccionan principalmente por
medio del mecanismo carbocatiónico.
En el párrafo inicial de esta sección se afirmó que la protonación era
el efecto catalítico más importante y el más simple de la química orgánica. En
presencia de ácidos, muchos tipos de átomos que se encuentran en compuestos
orgánicos se protonan significativamente: ixígeno, nitrógeno, azufre e incluso
carbono. Como veremos en casi todos los capítulos de este tecto, esta
protonación tiene efectos poderosos sobre reacciones de muchos tipos que
incluyen prácticamente toda clase de compuestos.
5.26 Análisis
de halogenuros de alquilo
Los halogenuros de alquilo simples responden a los ensayos comunes de caracterización
de la misma manera que los alcanos: son insolubles en ácido sulfúrico
concentrado y frío; inertes al bromo en tetracloruro de carbono, al
permanganato acuoso y al anhíbrido crómico. Sin embargo, se distinguen con
facilidad de los alcanos por medio del análisis cualitativo (Sec. 2.26), que
indica la presencia de halógeno.
En muchos casos, es posible detectar la presencia de halógeno sin la
oxidación de Schoniger ni la fusión con sodio. La muestra desconocida se
calienta durante algunos minutos con nitrato de plata alcohólico (el alcohol
disuelve tanto el reactivo iónico, como el compuesto orgánico): la formación de
un precipitado, insoluble en ácido nítrico diluido, indica la presencia de
halógeno.
Como en casi todas las
reaciones de halogenuros orgánicos, la reactividad frente al nitrato de plata alcohólico sigue la
secuencia RI >RBr >RCI. Para determinado átomos de halógeno, la
reactividad decrece en el orden 3º> 2º> 1º, la secuencia típica de la
formación de carbocationes. Más adelante, veremos que los halogenuros alílicos
(Sec. 10.13) y bencílicos (Sec. 15.18)
son muy reactivos. Otras pruebas (estereoquímica, transposiciones) señalan que
esta reacción es del tipo SN1. Se crece que el ión plata acelera la
reacción, arrancando el halogenuro del grupo alquilo.
![]()
(Los halogenuros de vinilo y arilo no reaccionan; Secs. 10.18 y 29.5.)
Como se mencionó antes (Sec. 5.3), los halogenuros sustituidos también
experimentan las reacciones características de sus otros grupos funcionales.
(El análisis espectroscópico de los halogenuros de alquilo se tratará
en el Cap. 16.)