Estereoquímica I.
Estereoisómeros
(Ir a página principal http://organica1.org/ )
4.1 Estereoquímica y estereoisomería
4.2 Números de isómeros y carbono tetraédrico
4.3 Actividad óptica, Luz polarizada en un plano
4.6 Enantiomería: el descubrimiento
4.7 Enantiomería y carbono tetraédrico
4.8 Enantiomería y actividad óptica
4.9 Pronósticos de la enantiomería. Quiralidad
4.13 Actividad óptica: un estudio más detallado
4.15 Especificación de la configuración: R y S
4.19
Especificación de la configuración: más de un centro quiral
4.20 Isómeros conformacionales
4.21 Reacciones que involucran estereoisómeros
4.22 Generación de un centro quiral. Síntesis y actividad
óptica
4.23
Reacciones de moléculas quirales. Ruptura de enlaces
4.24 Reacciones de moléculas quirales.
4.26 Reacciones de moléculas quirales.
4.27 Reacciones de moléculas quirales con reactivos ópticamente
activos. Resolución
4.28 Reacciones de moléculas quirales.
4.1
Estereoquímica y estereoisomería
La ciencia de la química orgánica, como hemos dicho, se
basa en la relación entre estructura molecular y propiedades. Aquella parte de la
ciencia que se ocupa de la estructura en tres dimensiones se denomina estereoquímica (del griego stereos, <<sólido>>).
Un aspecto de la estereoquímica es la estereoisomería. Recordemos que los isómeros son compuestos diferentes que tienen la misma
fórmula molecular.
La clase particular de isómeros que sólo se diferencian
por la orientación espacial de sus átomos (pero que son iguales entre sí en
cuanto a qué átomos están unidos a cuáles otros) se llama estereoisómeros.
Existen pares de estereoisómeros que se diferencian tan
poco estructuralmente -y, por consiguiente, en sus propiedades - que de todas
las mediciones físicas que podamos efectuar, solamente una, que requiere de un
instrumento especial y de un tipo excepcional de luz, puede distinguirlos. Sin
embargo, a pesar de su gran similitud, la existencia de tales isómeros nos
proporciona una de nuestras sondas más sensibles para la exploración de
mecanismos de reacciones químicas; muy
a menudo, se selecciona una de estos isómeros para un estudio, no por que sea diferente de los compuestos
ordinarios en su química tridimensional, sino porque puede revelar lo que las sustancias corrientes ocultan y, nuevamente, a pesar de su gran semejanza,
un isómero de tal pareja puede
servir de alimento nutriente, como
antibiótico o como un poderoso estimulante cardiaco, mientras que el otro puede
ser inútil.
Ya hemos comenzado nuestro estudio de la rama de la
estereoquímica llamada análisis conformacional (Secs. 3.3 y 3.5). En
este capítulo aprenderemos a pronosticar
la existencia de la clase de
estereoisómeros conocidos como enantiómeros
y diastereómeros, a representar y designar sus estructuras y, en forma general, a comparar sus propiedades. Luego, en la parte final
del capítulo, se destacará lo que estos isómeros son, cómo se forman, qué
hacen y qué nos pueden revelar. Pero la estereoquímica está omnipresente en
la química orgánica, por lo que
volveremos a ella una y otra vez a lo largo del libro, tanto para añadir a nuestros conocimientos los conceptos fundamentales de
la estereoquímica, como para utilizarla, con el fin de comprender mejor lo que
sucede en las reacciones químicas.
4.2 Números
de isómeros y carbono tetraédrico
Comencemos el estudio de la estereoquímica con el metano
y algunos productos de sustitución sencillos. Todo compuesto, por complejo que
sea, que tenga carbono unido a otros cuatro átomos, puede ser considerado como
un derivado del metano; y todo lo que
aprendemos acerca de la forma de su molécula puede ser aplicado a formas de
moléculas mucho más complejas.
La espectroscopia, las difracciones de rayos X y la
electrónica muestran que cuando los enlaces del carbono están unidos a otros cuatro átomos, sus
enlaces están dirigidos hacia los
vértices de un tetraedro. Sin embargo, ya en 1874, muchos años antes de que fuera posible la determinación directa de la estructura
molecular, J. H. Van’t Hoff (todavía estudiante de la Universidad de Utrecht), y por otra parte, J. A. LeBel,
propusieron el átomo de carbono tetraédrico. Su proposición se basaba en la prueba del número de isómeros.
Para cualquier átomo Y, nunca se ha encontrado más
de una sustancia de fórmula CH3Y:
la cloración del metano solamente da un compuesto de fórmula CH3CI;
la bromación da un solo CH3Br. Análogamente, sólo se conoce un CH3F y un solo CH3I;
es más, lo mismo es cierto si Y no sólo representa un átomo,
sino a un grupo de ellos (al menos que el grupo sea tan complicado que genere
isomería por sí mismo): solamente existe un CH3OH, un solo CH3COOH,
un solo CH3SO3H.
¿Qué sugiere esto en cuanto a la distribución de los átomos del metano? Sugiere que todos los
hidrógenos del metano son equivalentes, de modo que la sustitución de uno daría
un producto distinto que la sustitución de otro, con lo que se obtendrían
compuestos isómeros de sustitución.
¿En que forma pueden ordenarse los átomos del metano,
para que los cuatro hidrógenos resulten equivalentes? Hay tres arreglos
posibles: (a) uno plano (I), en el
que el carbono se encuentra en el centro de un rectángulo (o cuadrado) y un
hidrógeno en cada vértice; (b) una distribución piramidal (II), con el carbono en el ápice de una pirámide y un
hidrógeno en cada vértice de una base cuadrada; (c) un arreglo tetraédrico (III), con el carbono en el
centro de un tetraedro y un hidrógeno en cada uno de sus vértices.
¿Cómo podemos saber que cada uno de estos ordenamientos
sólo puede dar origen a una sustancia de fórmula CH3Y? Como siempre,
cuando se trata de este tipo de
problemas, la respuesta se encuentra al usar modelos moleculares. (Pueden
emplearse bolitas de goma y palillos para confeccionar estructuras como I y II, para las que no valen los ángulos de enlace
de los modelos moleculares corrientes.) Por ejemplo, construimos dos modelos
idénticos de I. Supongamos que en uno de ellos reemplazamos el H
superior por un átomo Y diferente,
representado por una bolita de color distinto; en el otro sustituimos, digamos,
el H inferior derecho. A continuación,
probamos si los modelos resultantes son superponibles; es decir, vemos si podemos hacerlos coincidir en
todas sus partes, para lo que se permite toda manipulación, excepto doblar o
quebrar enlaces. Si ambos son superponibles,
simplemente representan dos moléculas del mismo compuesto; si no lo son, representan moléculas de
compuestos diferentes, que son isómeros por definición (Sec. 1.24),
puesto que tienen la misma fórmula molecular. Cualquiera que sea el hidrógeno que reemplazamos en I (o en II
o en IIII), siempre obtenemos la
misma estructura. Para cualquier
arreglo diferente al de estos tres, obtendríamos más de una estructura.
En lo que concierne
a compuestos de fórmula CH3Y, la prueba del número de
isómeros limita la estructura del metano a una de estas tres posibilidades.
Para
todo átomo Y todo átomo Z, solamente se
conoce una sustancia de fórmula CH2YZ; por ejemplo, la halogenación del metano da
sólo un compuesto de fórmula CH2CI2, un solo CH2Br2
y un solo CH2ClBr.
De las tres posibles
estructuras del metano, solamente la tetraédrica es concordante con
estas pruebas.
Así, para el metano sólo la estructura tetraédrica
concuerda con la prueba del número de isómeros. Es cierto que esta evidencia es
negativa; puede argumentarse que
existen isómeros que nunca han sido
aislados o detectados sencillamente, porque las técnicas experimentales no son suficientemente
buenas; pero, como ya se ha dicho, todo compuesto que tiene carbono unido a otros cuatro átomos puede considerarse
como un derivado del metano; en
la preparación de cientos de miles de sustancias de este tipo, el número
de isómeros obtenidos siempre ha
concordado con el concepto del átomo de carbono tetraédrico.
Hay evidencias positivas adicional para el carbono
tetraédrico: el descubrimiento del tipo preciso de isómeros enantiómeros vaticinado para compuestos
de fórmula CWXYZ. La existencia de enantiómeros fue lo que convenció a Van’t
Hoff y LeBel de que el carbono es tetraédrico; pero para comprender lo que son los enatiómeros, debemos conocer
previamente la probabilidad llamada actividad
óptica.
4.3 Actividad
óptica, Luz polarizada en un plano
La luz posee ciertas propiedades que se comprenden mejor si se considera como un fenómeno ondulatorio, cuyas
vibraciones son perpendiculares a la dirección de su desplazamiento. Hay un
número infinito de planos que pasan por
la línea de propagación y la luz
ordinaria vibra en todos estos planos. Consideremos que se está mirando de frente una linterna, la
figura 4.1 muestra esquemáticamente el tipo de vibraciones que tienen
lugar, todas ellas perpendiculares a una línea entre nuestros ojos y el papel (linterna). La luz polarizada en un plano
es luz cuyas vibraciones ocurren en uno solo de sus planos posibles. La luz ordinaria se convierte en polarizada haciéndola pasar a través de una lente hecha
del material conocido como Polaroid o, más tradicional, por trozos de calcita
(una forma cristalina particular del CaCO3), dispuestos de forma que
constituyen lo que se conoce como un prisma
de Nicol.

Fig. 4.1 Representación esquemática de (a) luz ordinaria y (b) luz polarizada en un plano. La luz se propaga perpendicularmente a la página; las vibraciones están en el plano de la página.
Una sustancia
ópticamente activa es la que rota el
plano de la luz polarizada. Cuando se hace pasar luz polarizada, vibrando
en un plano determinado, por una sustancia ópticamente activa, emerge vibrando
en un plano diferente.
4.4 El
polarímetro
¿Cómo puede detectarse
esta rotación del plano de la luz polarizada, esta actividad óptica? Se
detecta y mide por medio de un instrumento
llamado polarímetro,
representado esquemáticamente en la figura 4.2. Consta de una fuente luminosa,
dos lentes (Polaroid o Nicol), y entre ellas
un tubo portador de la sustancia que se va a examinar para determinar su actividad óptica. La
disposición de estas piezas es tal que
la luz pasa por una de las lentes (polarizador), luego por el tubo, después
por la segunda lente (analizador), y
finalmente llega al ojo. Si el tubo está vacío, observamos que el máximo de luz
alcanza al ojo cuando la disposición de ambas lentes es tal que dejan pasar luz que vibra en el mismo plano. Si
rotamos la lente más cercana al ojo, por ejemplo, observamos que la luz se
amortigua y alcanza un mínimo cuando la lente está perpendicular a su posición
original.
Ajustamos las lentes de modo que pase el máximo de
luz. (En la práctica, es más fácil
detectar un mínimo que un máximo; el principio es el mismo.) Luego colocamos en
el tubo la muestra que se desea analizar. Si la sustancia no afecta al plano de
polarización, la transmisión lumínica
sigue siendo máxima, y se dice que el compuesto es ópticamente inactivo. En cambio, si la sustancia desvía el plano de
polarización, debe rotarse la lente más cercana al ojo para ajustarla al nuevo
plano, si se quiere que la transmisión lumínica sea otra vez máxima; se dice
que el compuesto es ópticamente activo.
Si la rotación del plano y, por consiguiente, el giro de la lente es hacia la
derecha ( en el sentido de las manecillas del reloj), la sustancia, es dextrógira ( del latín dexter, <<derecho>>); Si la rotación es hacia la
izquierda (contraria a las manecillas del reloj), es levógira (del latín laevus,
<<izquierdo>).
No sólo
podemos determinar que el
compuesto ha girado el plano y en qué dirección, sino también la magnitud del giro, que es simplemente el
número de grados que debemos rotar la lente para ajustarla a la luz. Se
emplean los símbolos + y - para indicar
giros a derecha e izquierda, respectivamente.
El ácido láctico (Sec. 4.7),
que se extrae del tejido muscular, gira la luz hacia la derecha, por lo que se
conoce como ácido láctico dextrógiro, o ácido (+)-láctico. El
2-metil-1-butanol que se obtiene del aceite de fusel (subproducto de la
fermentación del almidón a alcohol etílico), desvía la luz hacia la izquierda,
por lo que se le conoce como 2-metil-1-butanol
levógiro, o
(-)-2-metil-1-butanol.
4.5 Rotación
específica
Puesto que la rotación óptica del tipo que nos interesa
es causada por moléculas individuales del compuesto activo, la magnitud de la rotación depende de
cuántas moléculas sean interceptadas por la luz a su paso por el tubo.
En un tubo de 20 cm de largo, la luz se topará con el
doble de moléculas que en uno de sólo 10 cm, por lo que la rotación también
será doble. Si el compuesto activo se halla en solución, la cantidad de
moléculas con que se encuentra la luz depende de la concentración. Para un tubo de longitud dada, la luz
interceptará dos veces más moléculas en una solución de 2 g por 100 ml de
disolvente que en una con 1 g por 100 ml de disolvente, por lo que la rotación
será doble. Si se consideran la longitud del tubo y la concentración, resulta
que la magnitud del giro, además de su
sentido, es una característica de cada compuesto activo individual.
Rotación
específica es el número observado de grados de rotación si se
emplea un tubo de 1 Dm. (10 cm) de largo y si el compuesto examinado está
presente en la cantidad de 1 g/ml.
Para tubos de otras longitudes y concentraciones
diferentes, se calcula por medio de la ecuación
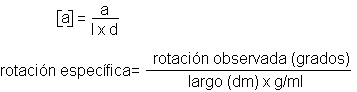
Donde d
representa la densidad de un líquido puro o la concentración de una solución.
La rotación específica es una propiedad tan
característica de un compuesto como lo es sus puntos de fusión y ebullición, su
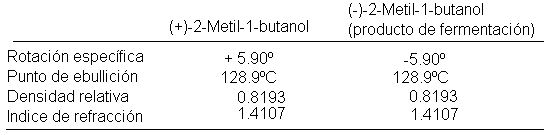
densidad o su índice de refracción. Así, la rotación específica del
2-metil-1-butanol obtenido del aceite de fusel es
![]()
Aquí, 20 corresponde a la temperatura y D a la longitud de onda de la luz empleada en la medición (línea D del sodio, 5893 Å).
4.6 Enantiomería:
el descubrimiento
La actividad óptica recién descrita fue descubierta en
1815 en el College de France por el físico Jean-Baptiste Biot.
En 1848, en la Ecole Normale de París, el químico Louis Pasteur hizo un conjunto de
observaciones que pocos años más tarde
le condujeron a formular una proposición que es la base de la
estereoquímica. Pasteur, en aquella época, aún hombre joven, había llegado a
la Ecole Normale del Colegio Real de Besancon (en el que recibió su baccalauréat és sciences con calificación de mediocre en química) recién doctorado en ciencias. Para adquirir
alguna experiencia en cristalografía,
estaba repitiendo el trabajo anterior de otro químico sobre sales del ácido
tartárico, cuando observó algo que nadie había
notado antes: el tartrato de sodio y amonio, ópticamente inactivo,
existía como una mezcla de dos clases diferentes de cristales, que
eran imágenes especulares entre sí.
Empleando una lupa y pinzas,
separó la mezcla cuidadosa y laboriosamente en dos montones minúsculos uno de cristales derechos y el
otro de izquierdos como quien separa guantes
izquierdos y derechos
desparramados sobre un mostrador
de tienda. Si bien la mezcla
original era ópticamente inactiva, cada
grupo de cristales, una vez disuelto en
agua, era ahora ópticamente activo.
Es más, las rotaciones específicas de ambas soluciones eran iguales, pero de signo contrario; es decir,
una solución rotaba la luz polarizada en un plano hacia la derecha y, la
otra, un número igual de grados hacia la izquierda. En todas las demás
propiedades, ambas sustancias eran
idénticas.
Puesto que la diferencia en rotación óptica fue observada
en solución, Pasteur concluyó que no
se trataba de características de los cristales, sino de las moléculas. Propuso
que, al igual que los dos tipos de cristales, las moléculas que los
conformaban eran imágenes especulares entre sí:
estaba proponiendo la existencia de isómeros, cuyas estructuras difieren sólo en que son imágenes
especulares y cuyas propiedades solamente difieren en la dirección de rotación de la luz polarizada.
Sólo faltaba que Van’t Hoff y LeBel señalaran que un
átomo de carbono tetraédrico, no sólo explicaría la ausencia de isómeros de fórmula CH3 Y CH2YZ, sino también la
existencia de isómeros especulares enantiómeros-,
como los ácidos tartáricos de Pasteur.
4.7 Enantiomería
y carbono tetraédrico
Convenzámonos de que, efectivamente deben existir tales
isómeros especulares. Partiendo de la verdadera disposición tetraédrica del
metano, construyamos un modelo de un
compuesto CWXYZ, empleando una esfera
de distinto color para cada átomo o
grupo diferente, representados por W,
X, Y Z. Luego, imaginemos que lo
colocamos frente a un espejo y construyamos un segundo modelo igual a la imagen
especular; tenemos ahora dos modelos con el aspecto siguiente:
que podemos representar, utilizando las fórmulas de cuña,
como sigue:

Como ya se ha visto (Fig. 2.2, Sec. 2.2), una cuña sólida
representa un enlace que sale del plano
del papel hacia nosotros, y una quebrada, un enlace que se aleja de nosotros
por detrás del plano del papel. (Una línea normal representaría un enlace en el
plano de papel.)
Ahora bien, ¿son superponibles estos modelos? No. Podemos
torcerlos y girarlos tanto queramos (mientras no se rompan los enlaces), pero
aunque pueden coincidir dos grupos de
cada uno, es imposible que lo hagan los otros dos. (Puede intentarse hace lo
mismo con las fórmulas de cuña.) Los modelos no son superponibles, por lo que
deben representar dos isómeros de fórmula CWXYZ.
Tal como se vaticinó, efectivamente existen isómeros especulares, y se conocen
miles de ejemplos, además de los ácidos tartáricos; por ejemplo, hay dos ácidos lácticos isómeros y dos 2-metil-1-butanoles,
dos ácidos cloroyodometanosulfónicos y dos cloruros de sec-butilo.
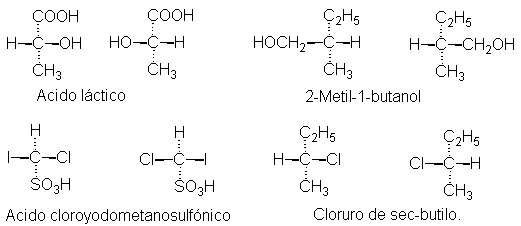
Podemos apreciar que las estructuras de cada par son
imágenes especulares; con modelos podemos verificar fácilmente que no son superponibles,
por lo que representan isómeros. (De hecho, ya lo hemos comprobado, puesto que
los modelos de CWXYZ que construimos
pueden, por supuesto, representar a cualquiera de ellos.)
Aún no necesitamos conocer la química de estas
sustancias, ni siquiera lo que representa una determinada colección de letras
(-COOH, por ejemplo, o -CH2OH); podemos distinguir cuándo ciertos
átomos o grupos son iguales o distintos, y si un modelo puede superponerse o no
a su imagen especular. Incluso dos isótopos, de un mismo elemento son
suficientemente diferentes, como el protio (hidrógeno ordinario, H) y el
deuterio (hidrógeno pesado, D) como para permitir isomería detectable:
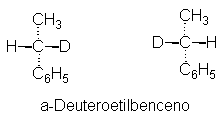
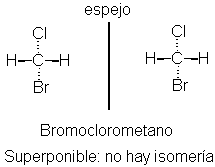
Debemos recordar que todo una imagen especular (salvo un
vampiro, desde luego), incluyendo todas las moléculas; la mayoría de ellas, sin
embargo, son superponibles a sus imágenes, como el bromoclorometano, por lo que
no exhiben esta isomería especular.
Los isómeros especulares
se llaman enatiómeros. Puesto que
sólo se distinguen por la diferente orientación espacial de sus átomos, los
enantiómeros pertenecen a la clase general
llamada estereoisómeros. Más
adelante nos encontramos con estereoisómeros que no son imágenes especulares entre sí, y se denominan diasterómeros. Cualquier par estereoisómero se clasifica como
enantiómero o como diasterómero, dependiendo de si son o no imágenes
especulares entre sí.
La imposibilidad de superposición de imágenes especulares
da origen a los enantiómeros y también,
como veremos, les confiere actividad óptica, por lo que a menudo se hace
referencia a ellos como (un tipo de ) isómeros
ópticos, término que no emplearemos, puesto que es difícil de definir en efectos, a menudo se usa sin definición y
de utilidad dudosa.
4.8 Enantiomería
y actividad óptica
La mayoría de los compuestos no rotan el plano de la luz
polarizada. ¿A qué se debe que algunos lo hagan? La causa no está en la familia
química específica a la cual pertenecen, puesto que se encuentran sustancias ópticamente
activas en todas ellas. Para descubrir
la característica estructural
especial que origina la actividad
óptica, examinemos más de cerca lo que
sucede al hacer pasar luz polarizada por un compuesto puro.
Cuando un haz de luz polarizada atraviesa una molécula
individual, casi siempre su plano se
rota ligeramente debido a la
interacción con las partículas cargadas de la molécula; la orientación y la
magnitud de la rotación varían con la
orientación de la molécula; particular
en el haz. En la mayoría de las sustancias, y debido a la distribución al azar
del enorme número de moléculas que
constituyen incluso la muestra más pequeña de un compuesto puro, por cada molécula que atraviesa la luz hay otra (idéntica) orientada como imagen especular de la primera, lo que cancela exactamente el
efecto. El resultado neto es la
ausencia de rotación, es decir,
la inactividad óptica, de modo que ésta no es una propiedad de las
moléculas individuales, sino más bien de la distribución
al azar de moléculas que pueden servir
de imágenes especulares recíprocas.
Por consiguiente, la inactividad óptica requiere que una
molécula de un compuesto actúe como imagen especular de otra; pero en el caso
especial de CWXYZ, descubrimos (Sección 4.7) una
molécula cuya imagen especular no es
simplemente otra molécula idéntica, sino una de un compuesto isómero diferente.
En una muestra de un enantiómero puro, ninguna molécula puede servir como
imagen especular de otra; no hay anulación de rotaciones, y el
resultado neto es la actividad óptica. Así, la misma imposibilidad de
suponer imágenes especulares que
origina la enantiomería es también la que explica la actividad óptica.
4.9 Pronósticos
de la enantiomería. Quiralidad
Las moléculas
no superponibles con sus imágenes especulares son quirales.
La quiralidad es condición necesaria y suficiente para la
existencia de los enantiómeros, es decir, un
compuesto cuyas moléculas son quirales
pueden existir como enantiómero; un
compuesto cuyas moléculas son aquirales ( sin quiralidad) no puede
existir como enantiómero.
Cuando decimos que una molécula y su imagen son
superponibles, significa que si para nuestro ojo mental sacáramos la imagen por
detrás del espejo donde parece estar,
podría hacerse coincidir en todas sus partes con la molécula. Por tanto, para
decidir si una molécula es quiral o no, hacemos unos modelos de ella y otro de
su imagen, y probamos si podemos superponerlos. Este es el modo más seguro,
puesto que si se maneja apropiadamente debe darnos la respuesta correcta. Es el método que debemos
emplear hasta habernos familiarizado bien con las ideas involucradas, e incluso es el método que deberíamos usar al encontrar un
compuesto nuevo.
Una vez familiarizados con los modelos, podemos
dibujarlos y tratar mentalmente de
superponerlos. Encontraremos que con algunos no es posible, como los
siguientes:
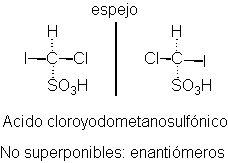
Estas moléculas son quirales y sabemos que el ácido cloroyodometanosulfónico puede
existir como enantiómero, con las
estructuras que acabamos de
construir o dibujar.
Otras resultan superponibles, como las siguientes:
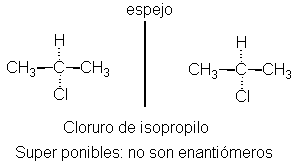
Estas moléculas son aquirales, por lo que sabemos que el cloruro
de isopropilo no puede existir como enantiómero.
<<Denomino quiral y digo que tiene quiralidad toda figura
geométrica, o todo grupo de puntos, si su imagen en su espejo plano,
idealmente realizada, no puede hacerse coincidir consigo misma. >> (Lord
Kelvin, 1893.)
En 1964, Cahn, Ingold y Prelog (véase
Sec. 4.15) propusieron que los químicos
emplearan los términos <<quiral>> y
<<quiralidad>> según la definición de Kelvin. Basada en la palabra
griega para <<mano>> (cheir),
quiralidad significa
<<sentido de las manos>>, refiriéndose a ese par de imágenes
especulares no superponibles que constantemente tenemos ante nosotros: nuestras
dos manos. Ha habido aceptación amplia de los términos de Kelvin, desplazando
en gran medida los términos más antiguos <<disimétrico>> y
<<disimetría>> (y los aún más antiguos y menos precisos
<<asimétrico>> y <<asimetría>>), aunque se encuentran
en la literatura química más antigua.
Como quiera que se denomine, la no superponibilidad con la imagen especular es condición necesaria y suficiente para la enantiomería; Es también condición necesaria pero no suficiente para la actividad óptica (véase Sec. 4.13).
4.10 El centro
quiral
Hasta este instante, todas las moléculas quirales descritas resaltan ser el tipo CWXYZ, es decir, en cada una de ellas hay un carbono (C*) que tiene cuatro grupos diferentes.
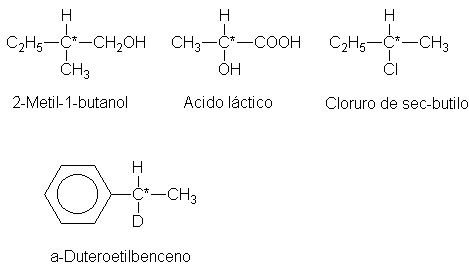
Un
átomo de carbono unido a cuatro grupos diferentes es un centro quiral. (A veces, se le llama carbono quiral, cuando se necesario
distinguirlo de nitrógeno quiral, fósforo
quiral, etc.)
Muchas moléculas pero
no todas que contienen un centro
quiral son quirales.
Muchas moléculas quirales pero no todas contienen un centro quiral. Hay moléculas con
centros quirales que, sin embargo, son aquirales (Sec. 4.18). (Tales moléculas aquirales contienen siempre más de un centro
quiral; si en una molécula sólo hay un centro quiral, podemos tener la
certeza que es quiral.) Hay moléculas
quirales que no tienen centros quirales (por ejemplo, véase Cap. 12, Problema 6).
La presencia o ausencia de un centro quiral no es, por
tanto, un criterio de quiralidad. Sin embargo, la mayoría de las moléculas quirales que estudiaremos
tienen centros quirales, por lo que
será útil buscarlos; si encontramos uno, debemos considerar la posibilidad de que dicha molécula sea
quiral, con lo que puede existir en forma de enantiómero. Más adelante (Sec. 4.18)
aprenderemos a reconocer la clase de molécula que puede ser aquiral, a pesar de
contener centros quirales; tales como moléculas tienen más de un centro quiral.
Una vez familiarizados con el empleo de modelos y
formulas de cuñas, podemos hacer uso de
representaciones aún más simples de moléculas con centros quirales, que pueden
dibujarse mucho más rápido. Es un método más peligroso, sin embargo, y debe aplicarse
apropiadamente para obtener respuestas exactas. Sencillamente trazamos
una cruz y agregamos a los extremos los
cuatro grupos unidos al centro quiral, que se
considera ubicado en el cruce de las líneas. Los químicos se han puesto de
acuerdo en que tal diagrama representa
una estructura específica: las líneas horizontales denotan enlaces que salen del
plano del papel hacia nosotros, mientras que las verticales indican uniones que
se alejan de nosotros por detrás del plano del papel; es decir:
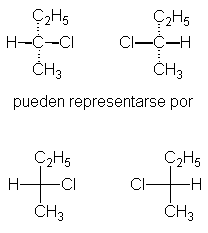
Al examinar la superponibilidad de estas dos representaciones
planas bidimensionales de objetos tridimensionales, debemos seguir cierto
procedimiento y obedecer reglas determinadas. En primer lugar, trazamos una de
ellas y luego la otra como su imagen especular. (Dibujar estas fórmulas al azar
puede conducir a conclusiones interesantes, pero bastante equivocadas con respecto al número de isómeros.) En tercer lugar,
podemos deslizar o rotar mentalmente
estas fórmulas en el plano del papel,
pero no podemos sacarlas de éste.
Estas representaciones son convenientes
si se usan con precaución: sin embargo, no son a prueba de tontos y, en casos dudosos, es preferible
emplear modelos o fórmulas de cuñas.
4.11 Enantiómeros
Los
isómeros que son imágenes especulares recíprocas se llaman enantiómeros. Los dos ácidos lácticos
cuyos modelos construimos en la sección 4.7 son
enantiómeros (del griego enantio <<opuesto>>);
También lo son los dos
2-metil-1-butanoles, los dos cloruros de sec-butilo, etc. ¿Cómo se
acojan las propiedades de los enantiómeros?
Los
enantiómeros tienen propiedades físicas idénticas, exceptuando la dirección de
rotación del plano de la luz polarizada; por ejemplo, ambos
2-metil-1-butanoles tienen idénticos puntos de fusión y ebullición, densidades,
índices de refracción y toda constante física que se pueda medir, excepto una:
uno rota la luz polarizada en un plano a la derecha, y el otro, a la izquierda,
hecho que no es sorprendente, puesto que las interacciones de ambos tipos de
moléculas con sus compañeras deben ser las mismas. Solamente la dirección de
la rotación es diferente; la magnitud es la misma, siendo la
rotación específica de uno de ellos +
5.90º, y la del otro, -5.90º. Al ser estas moléculas tan semejantes, resulta
razonable que puedan rotar la luz la misma magnitud. Son imágenes especulares,
como también lo son sus propiedades: la
imagen especular de una rotación en el sentido de las manecillas de un reloj es una rotación contraría y de,
exactamente, la misma magnitud.
Los
enantiómeros tiene propiedades químicas idénticas, excepto frente a reactivos
ópticamente activos. Los dos ácidos lácticos, no sólo son ácidos,
sino que, además, tienen la misma fuerza; es decir, disueltos en agua a
concentraciones iguales, ambos presentan el mismo grado de ionización. Los dos
2-metil-1-butanoles no sólo forman los mismos
productos alquenos por tratamiento con ácido sulfúrico caliente, bromuros de alquilo con HBr, ésteres con ácido acético, si no que también los forman
exactamente a la misma velocidad. Esto es bien razonable, puesto que los átomos
que sufren el ataque en cada caso se ven influidos en su reactividad por
exactamente la misma combinación de sustituyen tez. El reactivo que se acerca a
ambos tipos de molécula encuentra el mismo ambiente. Salvo, por supuesto, que
uno de ellos es la imagen especular del otro.
(Sólo hay un modo en que los enantiómeros pueden diferir
en sus reacciones con reactivos ordinarios, ópticamente inactivos: a veces dan productos que no son
idénticos, sino enantiómeros pero de todos modos, con la misma velocidad.
Veremos que puede ser muy significativo
si esto es así, o no, tanto desde un punto de vista práctico como teórico.)
En el caso especial de un reactivo que sea ópticamente
activo, por el contrario, las influencias ejercidas sobre él durante el ataque
a los enantiómeros no son idénticas, por lo que la velocidad de reacción será
diferente tan diferente en algunos casos que un isómeros no reacciona del todo.
Por ejemplo, en sistemas biológicos esta especialidad estereoquímica es más la
regla que la excepción, puesto que las enzimas,
los catalizadores de importancia capital y la mayoría de las sustancias sobre
las que actúan son ópticamente activas. El azúcar (+)-glucosa desempeña un papel
de gran importancia en el metabolismo
animal (Sec. 38.3) y es la base de una industria de fermentación
multimillonaria (Sec. 17.6); sin embargo, la
(-)-glucosa no es metabolizada por los animales, ni fermentada por las
levaduras. Cuando el moho penicillium glaucum se alimenta con una
mezcla de ácidos tartáricos enantiómeros, sólo consume el (+)-enantiómero,
dejando intacto al ácido (-)-tartárico. La actividad hormonal de la
(-)-adrenalina es muchas veces superior a la de su enantiómero; Sólo un
estereoisómero de la cloromicetina es un antibiótico. La (+)-efedrina no sólo
no tiene actividad como droga, sino
que, de hecho, interfiere en la acción de su enantiómero. Entre aminoácidos, la
asparagina y la leucina son dulces, y sólo un ácido glutámico aumenta el sabor
de los alimentos. El olor característico del aceite de menta verde se debe a la
(-)-carvona; pero la (+)-carvona es la esencia de la alcaravea.
Como analogía aproximada, comparemos una mano izquierda y
una derecha de igual fuerza (los enantiómeros), martillando un clavo (un
reactivo ópticamente inactivo) o, alternativamente, roscando un tornillo a derechas (un reactivo ópticamente activo).
Para clavar, ambas manos hacen uso de
un conjunto muscular exactamente
correspondiente, por lo que pueden
hacerlo a velocidades idénticas; en
cambio, para atornillar, emplean músculos diferentes: el pulgar derecho empuja,
por ejemplo, mientras que el izquierdo tira.
O bien, consideremos la reactividad del modo más preciso a nuestro alcance: por
medio del enfoque del estado de transición (Sec. 2.23).
Consideremos, en primer lugar, las reacciones de dos
enantiómeros con un reactivo ópticamente inactivo. En ambos casos, los
reactivos son de igual energía: un enantiómero, más el reactivo, y el otro, más
el reactivo. Los dos estados de transición de ambas reacciones son imágenes
especulares (son enantiómeros), por lo que también son de, exactamente, la
misma energía. En consecuencia, las diferencias en energías entre reactivos y
estados de transición los valores de las Eact-son idénticos lo mismo
que las velocidades de reacción.
Consideremos ahora las reacciones de dos enantiómeros con
un reactivo ópticamente activo. Nuevamente, los reactivos son de igual energía.
Sin embargo, lso dos estados de transición no son imágenes especulares (son
diastereómeros, Sec. 4.17), por lo que son
energéticamente diferentes: los
valores de las Eact son distintos, lo mismo que las velocidades de
reacción.
El principio subyacente en todo esto es: los enantiómeros
exhiben propiedades diferentes físicas o químicas solamente en un medio quiral. La luz polarizada proporciona un
medio así, y en él los enantiómeros difieren en una propiedad física: la
dirección de rotación de la luz. También pueden diferir en sus solubilidades en
un disolvente ópticamente activo, o en la absorción sobre una superficie
ópticamente activa. Para que los
enantiómeros reaccionen con velocidades distintas, se les puede
proporcionar el medio disolvente quiral o la superficie quiral de un catalizador;
Incluso para algunas reacciones catalizadas por luz mediante la irradiación con
luz polarizada circularmente. Para simplificar, utilizaremos a menudo el
término reactivo ópticamente activo o reactivo quiral al hablar de una reacción
sometida cualquiera de esas condiciones quirales. Emplearemos los términos
reactivo ópticamente inactivo o reactivo aquiral o, incluso, condiciones
ordinarias, al referirnos a una reacción en ausencia de un medio quiral.
4.12 Modificación
racémica
Una
mezcla de partes iguales de enatiómeros se denomina modificación racémica. Una modificación racémica es ópticamente
inactiva: Cuando se mezclan enantiómeros, la rotación provocada por una
molécula de un isómero cancelada exactamente por una rotación igual y opuesta
causada por una molécula de su enantiómero. Se emplea el prefijo ± para
especificar la naturaleza racémica de una muestra en particular, como, por
ejemplo, ácido (±)-láctico o (±)-2-metil-1-butanol.
Es útil comparar una modificación racémica con un compuesto
cuyas moléculas son superponibles sobre sus imágenes especulares; es decir, con
un compuesto aquiral. Ambas sustancias
son ópticamente inactivas exactamente por la misma razón. Debido a la distribución al azar de
un número muy grande de moléculas, por cada una de ellas que la luz atraviesa hay una segunda, imagen
especular de la primera, alineada de modo tal que cancela el efecto de la
primera. En una modificación racémica,
esta segunda molécula resulta ser un
isómero de la primera; Para un compuesto aquiral, no es un isómero, sino otra
molécula idéntica (Sec. 4.8).
(Hemos visto que para una sustancia ópticamente activa,
no contaminada con su enantiómero, la rotación no puede anularse, puesto que
ninguna molécula puede servir de imagen especular de otra, cualquiera que sea
el desorden de la distribución molecular.)
La identidad de la mayoría de las propiedades físicas de
los enantiómeros tiene una consecuencia de gran importancia práctica. No pueden
ser separados por métodos ordinarios: no es posible por destilación fraccionada, pues sus solubilidades en un
disolvente determinado son idénticas (a menos que éste sea ópticamente activo(;
ni por cromatografía, porque son retenidos
con igual firmeza por un
absorbente determinado (a menos que éste sea ópticamente activo). En
consecuencia, la separación de una modificación racémica en enantiómeros la resolución de una modificación
representa una tarea especial que debe ser abordada en forma muy particular (Sec. 4.27).
Por supuesto que la primera resolución fue la efectuada
por Pasteur con su lupa y pinzas (Sec. 4.6). Sin
embargo, este método es prácticamente inútil, puesto que las modificaciones
racémicas raras veces forman mezclas de
cristales reconocibles como imágenes especulares. De hecho, ni siquiera lo hace
el tartrato de sodio y amoniaco, a
menos que cristales a una temperatura
inferior a 28ºC, por lo que parte del mérito del descubrimiento de Pasteur ha sido atribuido al fresco clima parisino y, por supuesto, a la
disponibilidad de ácido tartárico procedente de los vinicultores franceses.
El método de resolución que casi siempre se emplea
también descubierto por Pasteur implica el uso de reactivos ópticamente activos
y se describe más adelante (Sec. 4.27).
Aunque su popularidad se debe principalmente a su gran
labor en bacteriología y medicina, Pasteur era químico, y su trabajo como tal
ya le hubiera asegurado una posición de gran científico.
4.13 Actividad óptica: un
estudio más detallado
Hemos visto (Sec. 4.8) que, al
igual que la enantiomería, la actividad óptica resulta de, y sólo de, la quiralidad:
la imposibilidad de superponer ciertas moléculas a sus imágenes especulares.
Cada vez que observamos actividad óptica (molecular), sabemos que estamos
tratando con moléculas quirales.
¿Es cierto lo contrario? ¿Debemos observar actividad
óptica cada vez que estamos en presencia de moléculas quirales, de compuestos que existen como enantiómeros? No.
Acabamos de ver que una mezcla de cantidades iguales de enantiómeros es
ópticamente inactiva; es evidente que, si hemos de observar actividad óptica, nuestro material debe tener un exceso de uno de los enantiómeros,
suficiente para que la rotación neta pueda ser detectada con polarímetro en
mano.
Además, el exceso de uno de los enantiómeros debe ser lo suficientemente persistente como
para alcanzar a medir la actividad. Si los enantiómeros sé interconvierten rápidamente, antes de poder medir la actividad debida a uno de ellos, se
obtuviese una mezcla en equilibrio, de composición 50:50 y ópticamente
inactiva, puesto que ambos enantiómeros son de la misma estabilidad
Aun cuando se cumplan todas estas condiciones, la
magnitud de la rotación óptica y en consecuencia su detectabilidad depende de
la molécula específica implicada; por ejemplo, en el compuesto I, los cuatro
grupos unidos al centro quiral solamente se diferencian en la longitud de las
cadenas.
Se ha calculado que este compuesto debería tener la
pequeñísima específica de 0.00001º muy por debajo de los límites de
detectabilidad para cualquiera de lso polarímetros existentes. En 1965, se
prepararon muestras enantioméricamente puras de ambos isómeros de I (véase Cap.
35, Problema 20), cada una de las cuales resultó ópticamente inactiva.
A este nivel de nuestro estudio, el problema de la
interconversión no nos causará dificultades. Prácticamente todas las moléculas
quirales que estudiaremos en este libro se encuentran en uno de dos extremos y
son fáciles de reconocer: (a) moléculas como las descritas en este capítulo que
deben su quiralidad a centros quirales; para éstas, la interconversión de enantiómeros
(enantiómeros configuracionales) es
tan lenta porque deben romperse enlaces
que su interconversión no tiene por qué preocuparnos. (B) Moléculas, cuyas formas enantiómeras (enantiómeros conformacionales) son interconvertibles
simplemente por rotaciones en torno a
enlaces simples; en este caso para los compuestos que veremos, la
interconversión es tan rápida que normalmente no necesitamos preocuparnos para
nada de la existencia de los enantiómeros.
4.14 Configuración
La disposición
de átomos que caracteriza a un estereoisómero determinado se llama configuración.
Empleando la prueba de superponibilidad, concluimos que hay dos cloruros de sec-butilo, por
ejemplo; sus configuraciones son I y
II. Supongamos que en laboratorio hemos obtenido muestras de dos compuestos de fórmula C2H5CHCICH3,
por métodos que estudiaremos más adelante (Sec.
4.27). Comprobamos que uno de ellos rota el plano de la luz polarizada hacia
la derecha, y el otro, hacia la izquierda; luego los colocamos en dos botellas,
una marcada <<cloruro de (+)-sec-butilo>>, y la otra,
<<cloruro de (-)-sec-butilo>>.
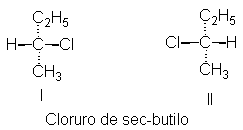
Hemos construido dos modelos para representar ambas
configuraciones de este cloruro y aislado dos compuestos isómeros con la
fórmula apropiada. Surge ahora una pregunta: ¿Cuál es la configuración de cada
isómero? El isómero (+), por ejemplo, ¿tiene la configuración I o la II? ¿Cómo saber cuál de las fórmulas
estructurales, I o II, hemos de poner
en cada botella? Es decir, ¿cómo asignamos
la configuración?
Hasta 1951, el problema de la configuración no tenía una
respuesta absoluta para ningún
compuesto ópticamente activo. En ese año, sin embargo, J. M. Bijvoet muy acertadamente director del Laboratorio Van’t
Hoff de la Universidad de Utrecht (Sec.
4.2)informó que había determinado la
disposición espacial verdadera de los átomos
de una sustancia ópticamente activa empleando un tipo especial de
análisis con rayos X (el método de la dispersión anómala). El compuesto era una
sal del ácido (+)-tartárico, el mismo ácido que casi exactamente 100 años antes
había conducido a Pasteur al descubrimiento de la isomería óptica.
Durante los años anteriores a 1951, las relaciones entre la configuración del
ácido (+)-tartárico y las de cientos de compuestos ópticamente activos ya
habían sido establecidas por métodos que se estudiarán más adelante (Sec. 4.24); Una vez conocida la configuración
del ácido (+)-tartárico, también quedaron establecidas de inmediato las otras.
(Para el caso de los cloruros de sec-butilo, por ejemplo, se sabe que el
isómero (-) tiene la configuración I, por lo que el (+) tiene la II.)
4.15 Especificación
de la configuración: R y S
Surge ahora otro problema. ¿De qué otro modo más sencillo
y conveniente podemos especificar una configuración determinada para no tener
que dibujarla cada vez? El método de utilidad más general emplea los prefijos R
y S, el cual implica dos pasos, de acuerdo con un procedimiento propuesto por
R. S. Cahn (The Chemical Society, Londres), sir
Christopher Ingold (University College, Londres) y V. Prelog (Eidgenossiche
Technische Hochschule, Zurich).
Paso 1.
Siguiendo un conjunto de reglas secuenciales
(Sec. 4.16), asignamos un orden de prioridad a
los cuatro átomos o grupos de átomos es decir, los cuatro ligandos unidos al
centro quiral.
En el caso del CHCIBrI, por ejemplo, los cuatro átomos
unidos al centro quiral son
todos diferentes y la prioridad
simplemente depende del número
atómico; el átomo de número mayor tiene preferencia. Así: I, Br, CI, H.
Paso 2.
Visualizamos la molécula orientada de
modo que el ligando de prioridad más baja se
aleje, y luego observamos el ordenamiento de los ligandos restantes. Si,
procediendo desde el ligando de
prioridad máxima hacia el de segunda y luego al de tercera, nuestra vista sigue
una trayectoria en el sentido de las manecillas del reloj, la configuración se especifica por R (del latín rectus, <<derecha>>); si lo
hace en el sentido contrario, es S (del latín sinister, <<izquierda>>).
Así, las configuraciones I y II se ven como sigue:
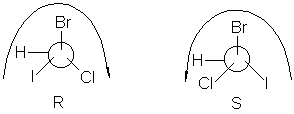
Y se especifican por R y S, respectivamente.
Para un compuesto ópticamente activo, un nombre completo
revela la configuración y la dirección de la rotación si es que son conocidas;
por ejemplo, cloruro de (S)-(+)-sec-butilo. Puede especificarse una
modificación racémica por el prefijo RS, por ejemplo, cloruro de (RS)-sec-butilo.
(La especificación de compuestos con más de un centro
quiral se estudia en Sec. 4.19.)
Desde luego, no debemos confundir la dirección de la rotación de una sustancia una propiedad física de un compuesto real, como su punto de fusión o de ebullición con la dirección que casualmente sigue nuestra vista al imaginarnos una molécula con una orientación arbitraria. En cuanto a nosotros, no tenemos idea de si la rotación (+) o (-) se asocia con la configuración R o la S, a menos que estemos informados de lo establecido experimentalmente para su compuesto específico.
4.16 Reglas
secuenciales
Para facilitar las referencias y la revisión, enunciaremos
aquí aquellas reglas secuenciales que necesitaremos. El lector debería estudiar las reglas 1 y 2 ahora, dejando la regla 3 para la ocasión en que surja la
necesidad de emplearla.
Regla
secuencial 1. Si los cuatro átomos unidos al centro quiral son todos
diferentes, la prioridad depende del número atómico; el que tenga mayor número
atómico tiene la prioridad más alta. Si dos átomos del mismo elemento, tendrá
mayor prioridad el de número de masa más alto.
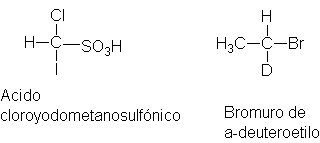
Así, en el caso del ácido cloroyodometanosulfónico, por
ejemplo, la secuencia es I, CI, S, H; en el bromuro de a-deuteroetilo es Br, C,
D, H.
Regla
secuencial 2. Si la regla 1 no puede decidir la prioridad relativa de
dos grupos, ésta será determinada por una comparación similar de los átomos
siguientes en cada grupo (y así sucesivamente, si fuera necesario, trabajando
desde el centro quiral hacia afuera).
Es decir, si dos átomos unidos al centro quiral son iguales, comparamos los
átomos unidos a cada uno de aquellos.
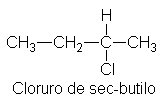
Como ejemplo, tomemos el cloruro de sec-butilo, en el
cual dos de los átomos unidos al centro quiral son carbonos. En el CH3,
los segundos átomos son H, H, H; en el C2H5 son C, H, H.
Puesto que el carbono tiene un número atómico mayor que el hidrógeno, C2,
H5 tiene mayor prioridad. Por consiguiente, la secuencia completa de
prioridad para el cloruro de sec-butilo es CI, C2H5, CH3,
H.
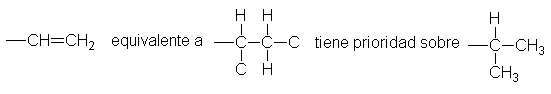
En el 3-cloro-2-metilpentano el C, C, H del isopropilo tiene prioridad sobre el C, H, H del etilo, de modo que la secuencia completa es CI, isopropilo, etilo, H.
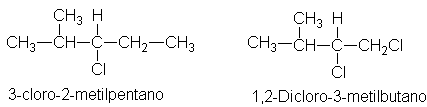
En el 1,2-dicloro-3-metilbutano el CI, H, H del CH2CI
tiene prioridad sobre el C, C, H del
isopropilo. El cloro tiene un número atómico mayor que el carbono, y el hecho
de que haya dos C y solamente un CI no influye. (Un número mayor vale más que
dos o tres de un número menor.)
Regla
secuencia 3. (El lector puede
aplazar el estudio de esta regla hasta que la necesite.)
Cuando aparece un doble o triple enlace, ambos
átomos se consideran como duplicados o
triplicados. Así,
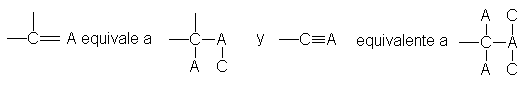
En el aldehído glicérico, por ejemplo, el grupo OH tiene
la prioridad más alta de todas,
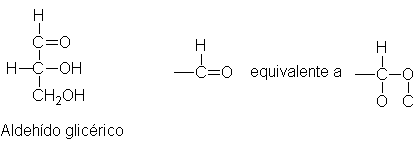
Y el O, O, H del CHO tiene preferencia sobre el O, H, H
del CH2OH. Luego la secuencia completa es OH, CHO, CH2OH,
H.
El grupo fenilo, C6H5, se trata
como si tuviera una de las estructuras
de Kekulé:
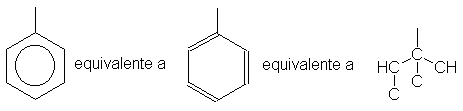
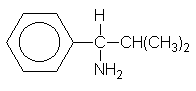
En el 1-amino-2-metil-1-fenilpropano, por ejemplo, el C,
C, C del fenilo tiene prioridad sobre el
C, C, H del isopropilo, pero no sobre el N, qué tiene un número atómico mayor. Por consiguiente, la secuencia
completa es NH2, C6H5, C3H7,
H.
El grupo vinilo, CH2=CH3, tiene
prioridad sobre el isopropilo.
Siguiendo la rama <<paterna>>, CH2-C,
en el vinilo llegamos a C, en vez del H en el CH2-H del isopropilo.
4.17 Diastereómeros
A continuación, debemos aprender a reconocer cuántos
estereoisómeros pueden existir para compuestos cuyas moléculas contienen no un
centro quiral, sino más de uno. (En
el Cap. 38, trataremos regularmente con moléculas que tienen cinco centros
quirales.)
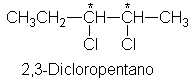
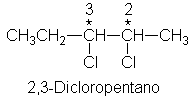
Comencemos con el 2,3-dicloropentano. Este compuesto
contiene dos centros quirales, C-2 y C-3. (¿Cuáles son los cuatro grupos unidos
a cada uno de estos átomos de carbono?)
¿Cuántos estereoisómeros son posibles?
Empleando modelos, construyamos primero la estructura I y
su imagen especular II, y veamos si son superponibles. Resulta que no lo son,
por lo que deben ser enantiómeros. Como antes, podemos representar estas
estructuras por fórmulas de cuña y tratar mentalmente de superponerlas, o usar
las representaciones de cruces, teniendo cuidado de no sacarlas del plano del
papel (Sec. 4.10).
A continuación, tratamos de interconvertir I y II por
medio de rotaciones en torno a enlaces carbono-carbono. Descubriremos que no es
posible interconvertirlas de esta manera,
por lo que cada una de ellas es capaz de retener su identidad y, si está
separada de su imagen especular, de mostrar actividad óptica.
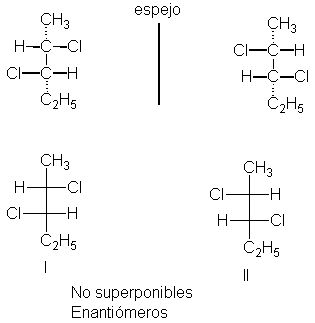
¿Existen otros estereoisómeros del 2,3-dicloropentano?
Podemos construir la estructura III, que resulta imposible de suponer a la
I y a la II; desde luego, no es la
imagen especular de ninguna de las dos. ¿Cuál es la relación entre III y I?
¿Entre III y II? Son estereoisómeros, pero no enantiómeros. Los estereoisómeros
que no son imágenes especulares entre
sí se denominan diastereómeros.
El compuesto III es un diastereoisómero de I y también de II.
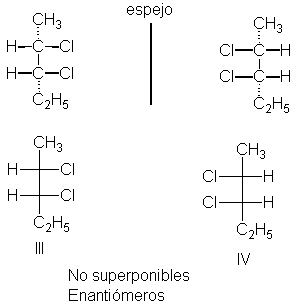
Ahora, ¿es III quiral? Empleando modelos, construimos su imagen especular, la estructura IV, y apreciamos que no es superponible (o interconvertible) con III, por lo que III y IV representan un segundo par de enantiómeros. Al igual que III, el compuesto IV es un diastereómero de I y II.
¿Cómo se comparan las propiedades de los diastereómeros?
Los
diastereómeros tienen propiedades químicas
similares, puesto que son miembros de la misma familia; sin embargo, esas propiedades no son idénticas. En la reacción de dos
diastereómeros con una sustancia determinada, ni los dos conjuntos de reactivos
ni los dos estados de transición son imágenes especulares, por lo que salvo por pura coincidencia no serán de
energías iguales. Los valores de las Eact serán diferentes, lo mismo
que las velocidades de reacción.
Los
diastereómeros tiene propiedades
físicas diferentes: Distintos puntos de fusión y de ebullición,
solubilidades en un disolvente determinado, índices de refracción, etc. También
difieren en la rotación específica: pueden tener igual o diferente signo de
rotación, y algunos ser inactivos.
Como consecuencia de sus diferencias en puntos de ebullición
y solubilidad, al menos en principio, pueden separarse por destilación o
cristalización fraccionadas; igualmente, debido a diferencias en forma
molecular y polaridad, difieren en
adsorción, por lo que pueden separarse por cromatografía.
Dada una mezcla de los cuatro 2,3-dicloropentanos,
podríamos separarla, por ejemplo, por destilación, en dos fracciones, pero no
en más: una de ellas sería la modificación racémica de I y II, mientras que la
otra sería la modificación racémica de III y IV. Una separación mayor
requeriría la resolución de ambas
modificaciones utilizando reactivos ópticamente activos (Sec. 4.27).
De este modo, la presencia de dos centros quirales pueden
conducir a la existencia de hasta cuatro estereoisómeros. Para compuestos que
tienen tres centros quirales podría
haber hasta ocho, estereoisómeros; para los que contienen cuatro, hasta dieciséis, y así sucesivamente. El número
máximo de estereoisómeros que pueden existir es igual a 2n, donde n
corresponde al número de centros quirales. (En todos los casos en los que
existen compuestos meso, hay menos de
este número máximo, como se verá en la próxima sección.)
El azúcar (+)-glucosa es, con mucho, el carbohidrato más
importante y abundante (Cap. 38). Se trata de la sustancia que se oxida en
nuestras células para proporcionar energía; es el principio que da origen al
almidón, del cual procede, en última instancia, nuestro alimento, y de la
celulosa, la armazón de las plantas que sintetizan el almidón. La glucosa
contiene cinco centros quirales, lo que podría dar origen y lo hace a 25 ó 32 estereoisómeros. Sólo uno de éstos, la a-D-glucosa, es la
unidad del almidón, y solo uno, la ß-D-glucosa, es la unidad de la celulosa.
4.18 Estructuras
meso
Observe ahora el 2,3-diclorobutano, que también tiene dos centros quirales. ¿Existe este compuesto también en cuatro formas estereoisómeras?
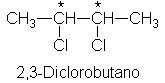
Empleando modelos como antes, llegamos primero a las dos estructuras V y VI, que son imágenes especulares no superponibles o interconvertibles; Por lo que son enantiómeros, y cada una de ellas debe ser ópticamente activa.
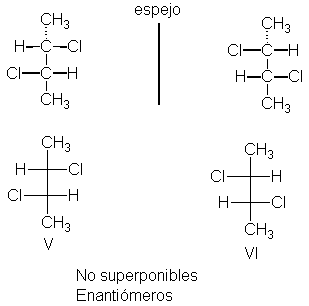
Luego construimos VII, que resulta ser un diastereómero de
V y VI, con lo que tenemos un total de
tres estereoisómeros. ¿Habrá un cuarto? No. Si construimos VIII, la imagen
especular de VII, resulta que ambos son superponibles; si giramos VII media
vuelta, coincide en todos los aspectos con VII. A pesar de sus centros
quirales, VII no es quiral: no puede existir en dos formas enantiómeras y no
puede ser ópticamente activo; se denomina compuesto meso.

Un compuesto meso
es aquel cuyas moléculas son superponibles
a sus imágenes especulares, a pesar de contener centros quirales. Un
compuesto meso es ópticamente inactivo por la misma razón que lo es otro cuyas
moléculas son aquirales: la rotación igual y puesta provocada por otra molécula que sea la imagen especular de
la primera (Sec. 4.8).
A menudo podemos reconocer una estructura meso a primera vista por el hecho de que una
mitad de la molécula es la imagen especular de la otra (al menos en una de sus conformaciones). Esto puede
apreciarse en el meso-2-3-diclorobutano,
si nos imaginamos la molécula dividida
por un plano situado donde está la línea de puntos. La molécula tiene un plano de simetría, por lo que no
puede ser quiral. (Advertencia: si no vemos un plano de simetría, por lo que no puede ser quiral. (Advertencia: Si no vemos un plano de simetría, esto no
significa necesariamente que la molécula sea quiral.)

4.19 Especificación de la configuración: más de un centro quiral
¿Cómo especificamos de compuestos que tiene más de un
centro químico? No presentan un problema especial, pues simplemente
especificamos la configuración en torno
a cada uno de los centros, y la numeramos
de acuerdo con el carbono al que corresponde.
Por ejemplo, consideremos los 2,3-dicloropentanos (Sec. 4.17). Tomamos los centros quirales, C-2 y
C-3, por separado ignorando la
existencia del otro, y seguimos los pasos de la sección 4.15, empleando las
reglas secuenciales de la sección 4.16.
El orden de prioridades de los cuatro
grupos unidos al C-2, es CI, CH3CH2CHCI,
CH3,H; para el C-3 es CI, CH3CHCI, CH3CH2,
H. (¿Por qué CH3CHCI tiene prioridad sobre CH3CH2
Tomando en nuestras manos o mentalmente un modelo del
estereoisómero particular que nos
interesa, concentramos nuestra atención primero sobre el C-2 (ignorando el C-3)
y luego sobre el C-3 (ignorando el C-2). El estereoisómero I (Sec. 4.17), por ejemplo, lo especificamos
(2S,3S)-2,3-dicloropentano; en forma análoga, II es (2R,3R), III es (2S,3R) y
IV es (2R,3S). Estas especificaciones nos ayudan en el análisis de las
relaciones entre estereómeros, I y III tienen configuraciones opuestas en un
centro quiral, y las mismas en torno al otro: 2S, 3S y 2S,3R.
Pueden manejarse el 2,3-diclorobutano (Sec. 4.18) de la misma forma. Sucede aquí que
ambos centros quirales ocupan posiciones equivalentes a lo largo de la cadena,
por lo que no es necesario usar números en las especificaciones. Los
enantiómeros V y VI (Sec. 4.18) se caracterizan
como (S,S)- y (R,R)-2,3-diclorobutano, respectivamente. El isómero meso, VII, puede, por supuesto,
especificarse como (R,S)- o como (S,R)-2,3-diclorobutano; la ausencia de
números enfatiza la equivalencia de ambas caracterizaciones. La relación
especular entre los dos extremos de la molécula es consecuente con las designaciones
R y S opuestas para dos centros
quirales. (Por supuesto, no todos los isómeros (R,S) son estructuras meso, son
solamente aquellos cuyas dos mitades son químicamente equivalentes.)
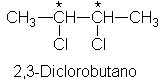
4.20 Isómeros
conformacionales
En la sección 3.5, vimos que hay varias conformaciones
escalonadas diferentes del n-butano, cada una de las cuales se encuentra en el
fondo de un valle energético en un mínimo
de energía y separada de las otras por cimas energéticas (véase Sec. 3.5,
Fig. 3.8). Las conformaciones diferentes
que corresponden a mínimos de energía se llaman isómeros conformacionales o confórmeros. Puesto que los isómeros
conformacionales solamente se diferencian entre sí por la orientación espacial
de sus átomos, también son estereoisómeros y, como tales, los pares de confórmeros pueden ser o no imágenes
especulares entre sí.
El n-butano existe en forma de tres isómeros
conformacionales: uno anti y dos oblicuos (Sec. 3.5). Los confórmeros oblicuos, II y III, son imágenes
especulares mutuas, por lo que son enantiómeros (conformacionales). Los
confórmeros I y II (o I y III) no son imágenes especulares, por lo que son
diastereómeros (conformacionales).
Aunque la barrera rotacional para el n-butanol es algo
mayor que para el etano, aún es suficientemente baja como para que al menos, a temperaturas ordinarias la interconversión de confórmeros
sea fácil y rápida . Existe un equilibrio que favorece la existencia de mayor
cantidad del confórmero anti, más
estable; las cantidades de ambos confórmeros
oblicuos son, desde luego, iguales,
puesto que son imágenes especulares y, por tanto, de estabilidad igual.
Expresado de otra manera, toda molécula pasa la mayor parte del tiempo en la
conformación anti, y el resto, se
encuentra a aprtes iguales en las dos oblicuas.
Como resultado de la rápida interconversión, estos isómeros no son separables.
La facilidad de interconversión es característica de casi
todo conjunto de isómeros conformacionales, propiedad por la cual
tales isómeros se diferencian más de la
clase de esteroisómeros que hemos estudiado en este capítulo. La diferencia en
interconvertibilidad se debe a la diferencia en al altura de la barrera energética
que separa los estereoisómeros, que, a su vez, resulta de un origen
diferente. Por definición la interconversión de isómeros conformacionales
implica rotación en torno a enlaces simples; la barrera rotacional es muy baja
en la mayoría de los casos, por lo que la interconversión es fácil y rápida. El
otro tipo de estereoisómeros, los isómeros
configuracionales o inversionales, difieren entre sí en configuración en
torno a un centro quiral. En este caso, la interconversión implica la ruptura
de un enlace covalente, para la que existe una barrera muy alta: 50 kcal/mol o
más (Sección 1.14). La interconversión es difícil y despreciablemente lenta, a
menos que de forma deliberada se den als condiciones para lograrla.
La interconvertibilidad de estereoisómeros es de
importancia práctica considerable, pues limita las posibilidades de su aislamiento. Los estereoisómeros
difíciles de interconvertir son separables (desde luego, por métodos
especiales, en el caso de la resolución de enantiómeros) y pueden estudiarse de
forma individual; entre otras cosas, se puede medir su actividad óptica. Los
isómeros fácilmente interconvertibles no son separables y los isómeros
idividuales aislados no pueden observarse actividad óptica, puesto que todas
las moléculas quirales sólo se encuentran como modificaciones racémicas que no
pueden resolverse.
En consecuencia, el estudio general de estereoisómeros
implica dos etapas; en primer lugar, ensayamos la superponibilidad de posibles estructuras isómeras, y luego
ensayamos su interconvertibilidad.
Ambas pruebas resultan mejor con modelos. Construimos modelos de las dos
moléculas y, sin permitir rotaciones en torno a enlaces simples, tratamos de
superponerlos; si esto no es posible, representan isómeros. A continuación,
permitimos todos los giros posibles de los modelos en torno a enlaces simples y
trataremos repetidamente de
superponerlos, si esto aún no es posible, no son interconvertibles, por lo que
representan isómeros configuracionales,
pero si después de las rotaciones son superponibles, son interconvertibles y
representan isómeros conformacionales.
Al tratar los aspectos de la estereoquímica que dependen
del aislamiento de estereoisómeros como número de isómeros o actividad óptica,
por ejemplo, o el estudio de las reacciones de un solo isómero, podemos ignorar
la existencia de isómeros fácilmente interconvertibles,
lo que significa la mayoría de los
isómeros conformacionales. Por conveniencia, <<la regla básica>>
siguiente específicamente otra cosa, los
términos <<estereoisómeros>>, <<enantiómeros>> y
<<diastereómeros>> sólo se referirán a isómeros configuarcionales,
incluyendo la geométricos (Sec. 7.6), y excluirán a los isómeros
conformacionales. Estos últimos se indicarán como <<isómeros
conformacionales>>, <<confórmeros>>, <<enantiómeros
conformacionales>> y <<diastereómeros conformacionales>>.
Entre estereoisómeros fácil y difícilmente
inconvertibles, no hay límite preciso; aunque podemos asegurar que la
interconversión de isómeros configuracionales será fácil, no podemos afirmar lo
mismo para los isómeros conformacionales. Según sea la naturaleza y el tamaño
de los sustituyentes, la barrera rotacional de enlaces simples puede tener
cualquier altura, desde la baja del etano, hasta una comparable a la ruptura de
un enlace covalente. Existen algunos isómeros conformacionales que pueden
aislarse, conservarse y estudiarse fácilmente; en efecto, el estudio de tales
isómeros (atropisómeros) es una parte
extensa y sumamente importante de la estereoquímica que, lamentablemente, no
podemos presentar en este libro introductorio. También existen otros isómeros
conformacionales aislables, no a temperaturas ordinarias, sino a temperaturas
más bajas, a las cuales la energía media de las colisiones es menor. Sin
embargo, los isómeros conformacionales que encontramos en este libro tienen
barreras rotacionales bajas, por lo que podemos suponer que mientras no
tengamos información en contrario cuando clasificamos estereoisómeros como
configuracionales o conformacionales, estamos clasificándolos como fácil o
difícilmente interconvertibles.
4.21 Reacciones
que involucran estereoisómeros
Hasta este momento, el estudio de la estereoquímica se ha
limitado principalmente a encontrar cuáles son los diversos tipos de
estereoisómeros, cómo diagnosticar su existencia y cómo nombrarlos y
clasificarlos. Hemos comparado sus propiedades, pero sólo de manera, muy
general.
Partamos ahora de la existencia
de estereoisómeros y ocupémonos de su participación en las reacciones químicas:
reacciones en las que se forman
estereoisómeros y reacciones en las que se consumen;
reacciones en las que el reactivo es del tipo corriente (es decir, ópticamente
inactivo) y aquellas en las que es ópticamente activo.
Estudiaremos:
(a)
La conversión de una molécula aquiral en una quiral, con
generación de un centro quiral;
(b)
(B) reacciones de moléculas quirales en las que no se
rompen enlaces con el centro quiral, y
ver cómo pueden usarse estas reacciones para relacionar la configuración de un
compuesto con la de otro;
(c)
(C) reacciones como las de (b), en las que se genera un
segundo centro quiral;
(d)
(D) reacciones de compuestos quirales con reactivos
ópticamente activos.
Luego examinaremos la estereoquímica de una reacción que
ya hemos estudiado la halogenación de alcanos por radicales libres y veremos
cómo puede utilizarse la estereoquímica para obtener información acerca del mecanismo de la reacción. Al hacer esto,
analizaremos:
(e)
Una reacción de un compuesto quiral, en las que se rompe
un enlace con un centro quiral.
4.22 Generación
de un centro quiral. Síntesis y actividad óptica
Uno de los productos de la cloración del n-butano es el compuesto quiral cloruro
de sec-butilo, que puede existir como dos enantiómeros, I y II, que se
especifican como S y R, respectivamente (Sec. 4.16).
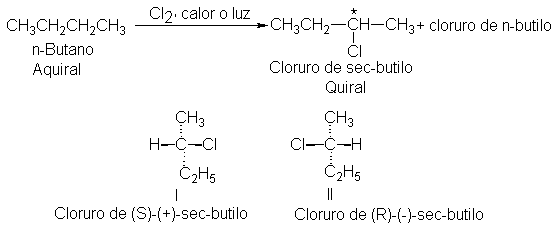
Cada enantiómero debe ser ópticamente activo. Si
colocáramos en el polarímetro el
cloruro de sec-butilo, preparado por
cloración del n-butano, ¿rotaría el
plano de la luz polarizada? La respuesta es no,
porque si se preparó según lo descrito, se obtendría la modificación racémica.
La siguiente pregunta es: ¿Por qué se
forma la modificación racémica?
En la primera etapa de la reacción, el cloro extrae
hidrógeno para dar cloruro de hidrógeno y un radical libre sec-butilo. El carbono del radical libre que tiene el electrón
impar presenta hibridación sp2 (trigonal,
Sec. 2.22), por lo que parte de la molécula es plana, y el carbono trigonal y los tres átomos unidos a él se
hallan en el mismo plano. En el segundo paso, el radical libre extrae cloro de
una molécula de este elemento para dar cloruro de sec-butilo. Pero el cloro puede unirse al radical plano por ambas
caras y, según cuál sea la cara,
resulta uno de dos productos: R o S (véase Fig. 4.3).
Puesto que la probabilidad de conectarse a una de las caras es exactamente
igual que para la otra, se obtienen los enantiómeros en cantidades idénticas;
el producto es la modificación racémica.
Si aplicáramos lo que acabamos de estudiar a la síntesis
de un compuesto cualquiera basándonos en cualquier mecanismo, correcto o
incorrecto, siempre llegaríamos a la misma conclusión: mientras no sean
ópticamente activos el material de partida ni el reactivo (ni el ambiente), se
debería obtener un compuesto ópticamente inactivo. En algún paso de la
secuencia de reacciones habrá dos caminos alternativos, uno de los cuales da un
enantiómero, y el otro, el enantiómero contrario. Ambas alternativas serán
siempre equivalentes y su elección será el azar. Los hechos concuerdan con
estas predicciones: la síntesis de
compuestos quirales a partir de
reactivos aquirales siempre produce la modificación racémica. Este es
simplemente un aspecto de una regla más general: reactivos ópticamente inactivos generan productos también inactivos.
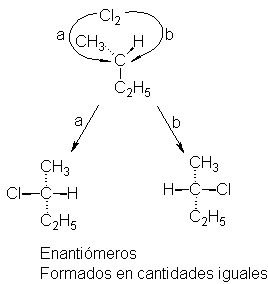
Fig. 4.3 Generación de un centro quiral. El cloro se une a una cara del radical libre plano, vía (a) o (b), para dar enantiómeros en cantidades iguales.
Para purificar el cloruro de sec-butilo obtenido por cloración del n-butano, llevaríamos a cabo
una destilación fraccionada. Sin embargo puesto que los cloruros de sec-butilo enantiómeros tienen
exactamente el mismo punto de ebullición, no son separables, recogiéndose ambos
en la misma fracción. Si se intentara la recristalización, tampoco habría
separación, ya que sus solubilidades son idénticas en todo disolvente
(ópticamente inactivo).
Es fácil apreciar entonces que, cada vez que se forma una modificación racémica en una
reacción, aislaremos (por métodos
ordinarios) una modificación racémica.
Si una síntesis normal produce una modificación racémica
y ésta no es separable por los métodos usuales de destilación, cristalización,
etc. ¿cómo sabemos que el producto obtenido es una modificación racémica? Es
ópticamente inactiva, ¿cómo sabemos entonces que realmente se trata de una
mezcla de dos sustancias ópticamente activas? La separación de
enantiómeros (llamada resolución) se
puede lograr por métodos especiales, que implican el uso de reactivos
ópticamente activos y se estudiarán más
adelante (Sec. 4.27).
4.23 Reacciones de moléculas quirales. Ruptura de enlaces
Habiendo hecho un compuesto quiral, el cloruro de sec-butilo,
veamos qué sucede si se le somete a una cloración por radicales libres. Se
forman varios diclorobutanos isómeros, que corresponden al ataque en varias
posiciones de la molécula. (Problema:
¿Cuáles son estos isómeros?)
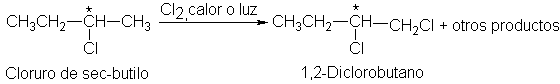
Por ejemplo, tomemos el cloruro de (S)-sec-butilo (que
rota la luz a la derecha, como vimos en Sec. 4.22)
y consideremos solamente aquella fracción de la reacción que produce
1,2-diclorobutano. Construyamos un modelo (I) de la molécula original,
empleando una sola esfera para C2H5 y esferas separadas
para cada átomo del CH3. Siguiendo los familiares pasos del
mecanismo, sacamos un H del CH3 y lo reemplazamos por Cl. Puesto que
en ninguno de los pasos hemos roto un enlace con el centro quiral, el modelo
que obtenemos necesariamente tendrá la configuración II, en la que la
disposición espacial en torno al centro quiral no ha cambiado o, en otras
palabras, se retiene la configuración, ocupando ahora un CH2Cl la
misma posición relativa que antes correspondía al CH3. Es un axioma
de la estereoquímica que las moléculas también se comportan de esta manera y
que una reacción que no involucra la
ruptura de un enlace con un centro quiral, procede con retención de la configuración
en torno a dicho centro.
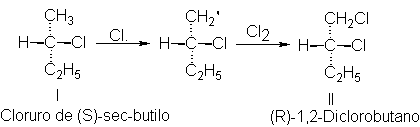
(Si una reacción procede con ruptura de un enlace con el centro quiral, no
podemos formular un pronunciamiento general acerca de la estereoquímica,
excepto que puede cambiar la configuración
y, probablemente, cambiará. Como se
estudiará en la Sec. 4.28, lo que suceda
dependerá del mecanismo de la reacción específica.)
Veamos ahora cómo se aplica el axioma sobre la ruptura de
enlaces para relacionar la configuración de un compuesto quiral con la de otro.
4.24 Reacciones
de moléculas quirales.
Relación de
configuraciones
Hemos estudiado (Sec. 4.14)
que la configuración de un enantiómero dado se puede determinar directamente
por un tipo especial de difracción de rayos X, utilizada por primera vez, en
1951, por BiJvoet, con el ácido (+)-tartárico. Sin embargo, el procedimiento es
difícil, consume mucho tiempo y sólo puede aplicarse a ciertos compuestos. No obstante
esta limitación, se conocen hoy día las configuraciones de cientos de
compuestos, que ya habían sido relacionados químicamente con el ácido
(+)-tartárico. La mayoría de estas relaciones fueron establecidas por
aplicación del axioma antes mencionado, es decir, la relación configuracional entre dos compuestos ópticamente activos
puede ser determinada por conversión de uno en el otro, mediante reacciones que
no impliquen la ruptura de enlaces de un centro quiral.
A modo de ejemplo, tomemos el (-)-2-metil-1-butanol (el
enantiómero que se encuentra en el aceite de fusel) y aceptamos momentáneamente
que tiene la configuración III, que especificaremos S. Convertimos este alcohol
en 1-cloro-2-metilbutano por reacción con el cloruro de hidrógeno. Aun sin conocer
el mecanismo de esta reacción, podemos darnos cuenta de que el enlace que se
rompe es el carbono-oxígeno. En torno al
centro quiral no se rompe ningún enlace, por lo que se retiene la configuración, ocupando un CH2CI
la misma posición relativa en el producto que ocupaba un CH2OH en el
reactivo. Colocamos el cloruro en un tubo y determinamos con el polarímetro que
el plano de la luz polarizada se rota a la derecha; es decir, el producto es
(+)-1-cloro-2-metilbutano. Puesto que (-)-2-metil-1-butanol tiene la
configuración III, (+)-1-cloro-2-metilbutano debe tener la configuración IV.
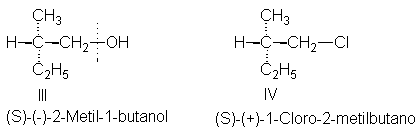
O bien oxidamos el (-)-2-metil-1-butanol con permanganato de potasio y obtenemos el ácido 2-metilbutanoico, el cual rota la luz hacia la derecha. Nuevamente, no se ha roto ningún enlace con el centro quiral, por lo que le asignamos la configuración V al ácido (+)-2-metilbutanoico.
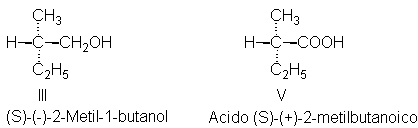
Casi siempre podemos decidir si se ha roto o no un enlace
con un centro quiral por simple inspección de las fórmulas del reactivo y del
producto, como hemos hecho en estos casos, y sin conocimiento del mecanismo de
la reacción. Sin embargo, debemos tener presente la posibilidad de ruptura y
reformación de un enlace en el transcurso de una reacción sin que esto se
observe en la superficie. Este tipo de cosas no sucede al azar, sino en ciertas
situaciones específicas que un químico orgánico aprende a reconocer.
De hecho, la estereoquímica tiene un papel preponderante
en el proceso de aprendizaje: uno de los mejores caminos para detectar estas
rupturas ocultas de enlaces es diseñado un experimento, que tiene que
involucrar un centro quiral si ha de suceder tal ruptura.
Pero, ¿cómo sabemos que (-)-2-metil-1-butanol tiene la
configuración III? Su configuración ha sido relacionada de la misma manera con
la de otro compuesto, y la de éste con la de otro más, y así sucesivamente,
hasta volver finalmente al ácido (+)-tartárico y al análisis por rayos X de Bijvoet.
Se dice que el (-)-2-metil-1-butanol, el (+)-cloruro y el
(+)-ácido tienen configuraciones similares (o iguales). Los enantiómeros de estas sustancias el (+)-alcohol, el
(-)-cloruro y el (-)-ácido forman otro conjunto de compuestos de configuración
similar. Se dice que el (-)-alcohol y el (-)-cloruro, por ejemplo, tienen
configuraciones opuestas. Veremos
que, generalmente, estamos más interesados en saber si dos compuestos tienen
configuraciones similares u opuestas que en conocer la configuración real de
cualquiera de ellos; es decir, nos interesan más las configuraciones relativas que las absolutas.
En este conjunto de compuestos con configuraciones
similares, observamos que dos son dextrógiros y el tercero es levógiro. El
signo de rotación es importante como medio para seguir la pista de un isómero
determinado, tal como podríamos usar el punto de ebullición o el índice de
refracción para distinguir si se tiene n-butano o isobutano, una vez asignadas sus configuraciones; pero el hecho de que dos
compuestos tengan igual u opuesto signo de rotación, tiene poco significado en
sí: pueden tener configuraciones similares o no.
Las tres sustancias han sido especificadas como S, pero
esto se debe a que, casualmente, CH2CI y COOH tienen la misma
prioridad relativa que CH2OH. Si reemplazáramos cloro por deuterio (Problema: ¿Cómo podría hacerse?), Se
especificaría el producto como R; Sin embargo, es evidente que tendría la misma
configuración que el alcohol, el halogenuro y el ácido. Si volvemos al cloruro
de sec-butilo y al 1,2-diclorobutano, observamos,
efectivamente, que las configuraciones similares I y II tienen especificación distinta, siendo uno S, y el otro, R; En este
caso, un grupo de prioridad inferior (-CH3) a la del –C2H5
se convierte en uno de prioridad mayor (-CH2CI). No podemos decidir
si dos compuestos tienen la misma configuración u opuesta por inspección de las
letras usadas para especificar sus configuraciones; debemos establecer y
comparar las configuraciones absolutas indicadas por dichas letras.
4.25 Pureza
óptica
Las reacciones en las que no se rompen enlaces con
centros quirales pueden emplearse para obtener otro tipo de información de gran
importancia: Las rotaciones específicas de compuestos ópticamente puros. Así,
por ejemplo, el 2-metil-1-butanol obtenido del aceite de fusel (que tiene una
rotación específica de 5,90º) es ópticamente
puro como la mayoría de las sustancias quirales de origen biológico; es
decir, se compone enteramente de uno de los enantiómeros, sin ninguna
contaminación de su imagen especular. Cuando se trata el material con cloruro
de hidrógeno, el 1-cloro-2-metilbutano obtenido resulta tener una rotación
específica de +1.67º. Puesto que no se ha roto ningún enlace con el centro
quiral, cada molécula de alcohol con configuración III es convertida en una de
cloruro con configuración IV; Como el primero era ópticamente puro, el segundo,
con rotación específica de +1,67º, también lo es. Una vez establecida esta rotación máxima, cualquiera puede
precisar la pureza óptica de una muestra del 1-cloro-2-metilbutano en unos
minutos con sólo medir su rotación específica.
Si una muestra del cloruro tiene una rotación de +0,835º,
sea, un 50% del máximo, se dice que tiene una pureza óptica del 50%. Consideramos que los componentes de la
mezcla son el isómero (+) y el (±); no el isómero (+) y el isómero (-). (Problema: ¿En qué porcentaje se
encuentran los isómeros (+) y (-) en esta muestra?)
4.26 Reacciones
de moléculas quirales.
Generación de un
segundo centro quiral
Volvamos a la reacción que sirvió de ejemplo en la sección 4.23, la cloración por radicales libres del
cloruro de sec-butilo,
concentrándonos esta vez en uno de los otros productos, uno en el que se genera
un segundo centro quiral: el 2,3-diclorobutano. Vimos (Sec.
4.18) que esta sustancia existe en tres formas estereoisómeras, una meso y dos enantiómeras.
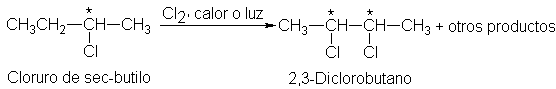
Supongamos que partimos del cloruro de sec-butilo
ópticamente activo (por ejemplo, el isómero S), efectuamos la cloración y
separamos los 2,3-diclorobutanos de los demás productos por destilación
fraccionada (o sea, de los isómeros 1,2-, 2,2-, etc.). ¿Qué estereoisómeros
puede contener nuestra muestra?
La figura 4.4 indica el
curso de la reacción. Se ilustran tres aspectos importantes:
(1)
Puesto que no se ha roto ningún enlace con el centro
quiral original, C-2, se retiene su configuración en todos los productos. Esto
es común a todos los casos en los que se genera un segundo centro quiral.
(2) Hay dos
configuraciones posibles para el nuevo centro quiral, C-3, y se forman ambas;
en este caso
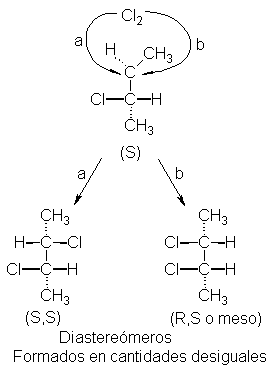
Fig. 4.4 Generación de un segundo centro quiral. La configuración
del centro quiral original no cambia. El cloro se combina según (a) o (b) para
dar diastereómeros en cantidades desiguales.
resultan de
los ataques (a) y (b) por lados opuestos de la parte plana del radical libre,
dando los
productos diastereómeros S,S y R,S (o meso).
En algunas
reacciones, pueden no generarse ambas configuraciones, pero siempre debemos
considerar la
posibilidad de que sí se generen; éste
es nuestro punto de partida. (Volveremos sobre este punto en la
Sec. 9.1.)
(3)
Los productos diastereómeros se generan en cantidades
distintas, en este caso porque el ataque (a) y el (b) no son igualmente
probables. Esto debe ser común a todos los casos en los que se generan
productos diastereómeros.
En la sección 4.22 vimos
que la generación del primer centro quiral da cantidades iguales de
enantiómeros; es decir, se forma la modificación racémica. Ahora observamos que
la generación de un segundo centro quiral en un compuesto que ya es ópticamente
activo produce una sustancia activa que contiene cantidades distintas de
diastereómeros.
Supongamos (como efectivamente es el caso) que los
productos del cloruro de (S)-sec-butilo
presentan una razón S,S:meso de
29:71. ¿Qué obtendríamos de la cloración de cloruro de (R)-sec-butilo?
Obtendríamos productos R,R y meso, y la razón R,R:meso sería exactamente 29:71. Cualquiera que sea el factor que
favorezca al producto meso a expensas del S,S, también favorecerá al producto meso sobre el R,R, y exactamente en el
mismo grado.
Finalmente, ¿Qué se espera obtener del cloruro de sec-butilo racémico ópticamente
inactivo? El isómero S presente daría los productos S,S y meso con una razón de
29:71; el isómero R formaría los compuestos R,R y meso, también en la razón 29:71. Puesto que hay exactamente la
misma cantidad de reactivos R y S, los dos conjuntos de productos se
equilibrarían recíprocamente, obteniéndose los productos racémico y meso en la razón 29:71; es decir, reactivos ópticamente
inactivos producen sustancias también inactivas.
Un aspecto requiere estudio adicional. ¿Por qué se forman
cantidades desiguales de diastereómeros? Porque el radical intermedio
3-cloro-2-butilo de la figura 4.4 ya contiene un
centro quiral, y carece de la simetría necesaria para el ataque sea igualmente
probable por ambas caras. (Constrúyanse un modelo del radical y asegúrese de
que esto es así.)
4.27 Reacciones
de moléculas quirales con reactivos ópticamente
activos. Resolución
Hasta el momento, sólo hemos estudiado las reacciones de
compuestos quirales con reactivos ópticamente inactivos; ahora nos ocuparemos
de reacciones con reactivos activos y examinaremos una de sus aplicaciones más
útiles: la resolución de una
modificación racémica; es decir, la
separación en enantiómeros de una modificación racémica.
Sabemos que cuando reactivos inactivos forman un
compuesto quiral, resulta una mezcla racémica (Sec.
4.22). Sabemos también que los enantiómeros que la constituyen tienen
propiedades físicas idénticas
(exceptuando la dirección de rotación de la luz polarizada), por lo que no
puede separarse por los medios usuales de destilación o cristalización
fraccionadas. Sin embargo, en este libro hay referencias frecuentes a
experimentos, realizados con el empleo de compuestos ópticamente activos, como el alcohol (+)-sec-butílico, el
(-)-2-bromo-octano, el cloruro de (-)-a-feniletilo, la (+)-a-fenilpropionamida.
¿Cómo se obtienen tales compuestos ópticamente activos?
Algunas sustancias ópticamente activas se obtienen de
fuentes naturales, dado que por lo
general los organismos vivos sólo producen un enantiómero del par. Así,
solamente se forma el (-)-2-metil-1-butanol en la fermentación de los almidones
con levadura, y sólo el ácido (+`)-láctico, CH3CHOHCOOH, en la
contratación muscular; de los juegos de frutas solamente se obtiene el ácido
(-)-málico, HOOCCH2CHOHCOOH;
y de la corteza de la cinchona sólo la (-)-quinina. De hecho, tratamos
con sustancias ópticamente activas en un grado cuya magnitud tal vez no
logramos imaginar. Nos alimentamos de pan y carne ópticamente activos; vivimos
en casas, usamos ropas y leemos libros, todo hecho de celulosa ópticamente
activa. Las proteínas que constituyen nuestros músculos y demás tejidos, el
glicógeno del hígado y de la sangre, las enzimas y hormonas que nos hacen
crecer y que regulan nuestros procesos metabólicos, son sustancias ópticamente
activas. Los compuestos naturales son ópticamente activos porque las enzimas
que provocan su formación y a menudo las materias primas de las que están
hechas son, a su vez, ópticamente activas.
Sólo podemos especular en cuanto al origen de las enzimas
ópticamente activas.
Se ha detectado la presencia de aminoácidos, las unidades
que constituyen las proteínas, en meteoritos, pero en cantidades tan pequeñas
que se ha especulado que <<lo que aparenta ser el golpeteo de pies
celestiales, probablemente sólo sea la huella digital de un pulgar terrenal>>.
Una parte de las pruebas de que los aminoácidos encontrados en un meteorito por
Cyril Ponnamperuna (Universidad de Maryland) son realmente de origen
extraterrestre, es el hecho de que son ópticamente inactivos, y no activos, como lo hubiesen sido contaminantes terrestres
procedentes de fuentes biológicas.
Con estos compuestos de origen natural se pueden hacer
otras sustancias ópticamente activas. Por ejemplo, hemos visto (Sec. 4.24) cómo se puede convertir el
(-)-2-metil-1-butanol en el cloruro o en el ácido correspondiente sin perder su
configuración; estos compuestos activos pueden convertirse, a su vez, en muchos
otros.
La mayoría de los compuestos activos se obtiene por
resolución de una modificación racémica, es decir, por separación de ésta en
sus enantiómeros. La mayoría de tales resoluciones se logran por medio de
reactivos que son ópticamente activos, que son generalmente de origen natural.
La mayor parte de las resoluciones efectuadas dependen de
la reacción de bases orgánicas con ácidos para dar sales. Por ejemplo,
supongamos que hemos preparado el ácido racémico (±)-HA. De varias plantas es
posible aislar ciertas bases muy complejas, llamadas alcaloides (es decir, semejantes a los álcalis), entre las que
figuran cocaína, morfina, estricnina y quinina. La mayoría de estos alcaloides
son productos por las plantas en sólo
una de dos formas enantiómeras posibles, por lo que son ópticamente activos. Tomemos una de estas bases, por ejemplo,
una levógira, (-)-B, y mezclémosla con nuestro ácido racémico ()-HA.
El ácido está presente en dos configuraciones, mientras
que la base solamente en una, por lo que resultarán cristales diferentes,
[(-)-BH+ (+)-A- y
(-)-BH+ (-)-A-].¿Cuál
es la relación entre estas dos sales? No son superponibles, puesto que no lo son
las partes ácidas. No son imágenes especulares, puesto que no lo son las partes
básicas; estas sales son estereoisómeros que no son enantiómeros, por lo que
son diastereómeros.
Estas sales diastereómeras tiene, por supuesto,
propiedades físicas diferentes, incluyendo la solubilidad en un disolvente
determinado, por lo que se pueden separar por cristalización fraccionada. Una
vez separadas ambas sales, se puede recuperar de ellas el ácido ópticamente
activo por adición de un ácido mineral fuerte, el cual desplaza al orgánico,
más débil. Si la sal ha sido purificada cuidadosamente para eliminar toda traza
de su diastereómero, entonces el ácido obtenido de ella es ópticamente puro. Entre los alcaloides que se suelen emplear para
este propósito figuran la (-) brucina, la (-)-quinina, la (-)-estricnina y la
(+)-cinconina.
La resolución de bases orgánicas se realiza invirtiendo
el procedimiento recién descrito: se usan ácidos ópticamente activos de origen
natural, como el ácido (-)-málico, por ejemplo.
La resolución de alcoholes, de especial importancia en
síntesis, presenta un problema especial: puesto que ni son apreciablemente
ácidos ni básicos, los alcoholes no pueden ser resueltos por formación directa
de sales, pero sí por medio de una adaptación bastante ingeniosa del método
recién descrito: se les conecta un <<mango>> ácido que permite la
formación de sales, y se elimina cuando ya no se necesite.
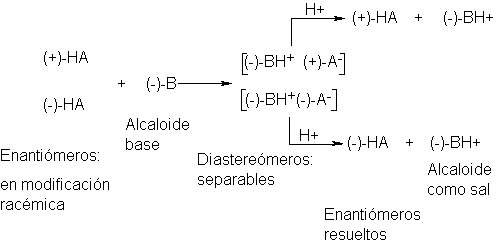
También pueden resolverse compuestos distintos de bases y
ácidos orgánicos o alcoholes. Aunque la química específica puede diferir de la
formación de sales descrita, el principio permanece igual: por medio de un reactivo ópticamente activo, una modificación
racémica se convierte en una mezcla de diastereómeros que se puede separar.
4.28 Reacciones
de moléculas quirales.
Mecanismo de la cloración por radicales libres
Hemos estudiado solamente reacciones de moléculas
quirales en las que no se rompen enlaces al centro quiral. ¿Cuál es la estereoquímica
de reacciones en las que se rompen enlaces al centro quiral?. La respuesta es: depende. Depende del mecanismo del proceso que se está
realizando; debido a esto, a menudo la estereoquímica puede proporcionarnos
información acerca de una reacción que no podríamos obtener de ninguna otra
forma.
Así, por ejemplo, la estereoquímica desempeñó un papel importante en el establecimiento del mecanismo que fue la base de todo nuestro estudio de la halogenación de alcanos (Cap. 3). Los pasos propagadores de este mecanismo son:
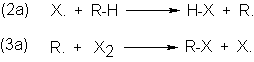
Hasta 1940, las pruebas existentes eran igualmente consecuentes con los pasos alternativos siguientes:
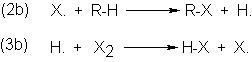
Para distinguir entre estos dos mecanismos, H. C. Brown,
M. S. Kharash y T. H. Chao (Universidad de Chicago) realizaron la halogenación
fotoquímica del (S)-(+)-1-cloro-2-metilbutano ópticamente activo. Obtuvieron,
desde luego, varios productos isómeros, correspondientes al ataque en diversos
lugares de la molécula. (Problema:
¿Cuáles eran estos productos?) Concentraron su atención en uno solo de ellos:
el 1,2-dicloro-2-metil-butano que resulta de la sustitución en el centro quiral
(C-2).
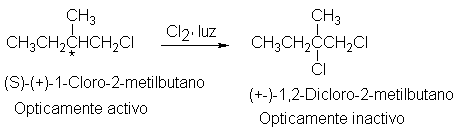
La base para la planificación del experimento fue la siguiente:
ambos mecanismos diferían en cuanto a si el radical libre alquílico es o no un
intermediario. Suponían que la estructura más probable para tal radical era plana lo que realmente es muy probable,
con lo que perdería su quiralidad original. El cloro se uniría con iguales
posibilidades a una u otra cara, con lo que se formaría un producto racémico
ópticamente inactivo; es decir, la reacción tendría lugar con racemización (véase Fig. 4.5).
Para el mecanismo alternativo, en el que el cloro se
uniría a la molécula mientras se
desplaza el hidrógeno, no podían formular ningún pronóstico, excepto que la
formación de un producto ópticamente inactivo sería altamente improbable: no
había ninguna razón para esperar que el ataque por detrás (en la cara opuesta al hidrógeno) tuviera lugar exactamente
en la misma proporción que por delante.
(En los desplazamientos iónicos, el ataque es, por lo general, por detrás.)
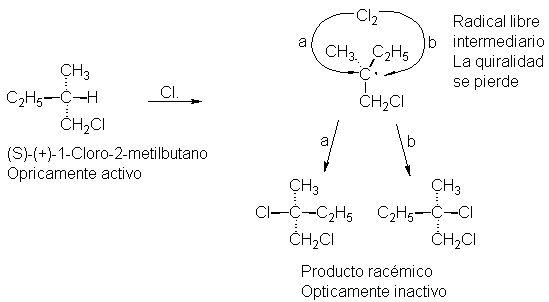
Fig. 4.5 Racemización por formación de radical libre. El cloruro se une a ambas caras del radical libre, según (a) o (b), para dar enantiómeros en cantidades iguales.
Por destilación fraccionada cuidadosa, separaron de la
mezcla de reacción el 1,2-dicloro-2-metilbutano y determinaron que era ópticamente inactivo, por lo que
concluyeron que el mecanismo que involucra radicales libres alquílicos, (2ª),
(3ª), es correcto. Este mecanismo se acepta ahora sin discusión: en la sección
2.21 vimos cómo las fuerzas relativas de los enlaces hidrógeno-cloro y
carbono-cloro fuerzan la reacción a que siga este curso.
Actualmente, se menciona el trabajo de Brown, Kharash y Chao
como prueba del comportamiento estereoquímico de radicales libres, con
inversión total del significado original del trabajo.
Estamos comenzando a apreciar que la estereoquímica
proporciona al químico orgánico una de sus armas más poderosas para descubrir
lo que sucede en una reacción.