Alcanos
(Ir a página principal http://organica1.org/ )
3.1 Clasificación por estructura: la familia
3.3 Libre rotación en torno al enlace simple carbono-carbono.
Conformaciones. Tensión torsional
3.5 Conformaciones del n-butano. Repulsión de Van der Waals
3.6 Alcanos superiores. La serie homóloga
3.9 Nombres comunes de los alcanos
3.11 Tipos de átomos de carbono e hidrógeno
3.14 Fuente industrial y preparación de laboratorio
3.16 El reactivo de Grignard: un compuesto organometálico
3.17 Acoplamiento de halogenuros de alquilo con
compuestos organometálicos
3.20 Mecanismo del halogenación
3.21 Orientación de la halogenación
3.22
Reactividades relativas de los alcanos en la halogenación
3.23 Facilidad de separación de átomos de hidrógeno. Energía
de activación
3.24 Estabilidad de radicales libres
3.25 Facilidad de formación de radicales libres
3.26 Estado de transición para la halogenación
3.27 Orientación y reactividad
3.28 Reactividad y selectividad
3.29 Ausencia de transposición en los radicales libres.
Trazadores isotópicos
3.32 Determinación de la estructura
Sustitución
por radicales libres.
3.1 Clasificación por estructura:
la familia
Hemos dicho que la base de la química orgánica es la
teoría estructural. Separamos todos los compuestos orgánicos en familias a
partir de sus estructuras. Una vez hecho esto, descubriremos que, al mismo
tiempo, hemos clasificado las sustancias de acuerdo con sus propiedades físicas
y químicas, de modo que una serie
específica de propiedades es característica de un tipo estructural en
particular.
Dentro de una familia hay variaciones en propiedades:
todos sus miembros pueden reaccionar con un reactivo específico, por ejemplo,
pero unos pueden hacerlo con mayor facilidad que otros. Puede haber diferencias
en un mismo compuesto, pudiendo ser una parte de su molécula más reactiva que
otra. Estas variaciones de propiedades corresponden a variaciones en las estructuras.
A medida que estudiemos las diversas familias de compuestos orgánicos, fijaremos primero
nuestra atención en su estructura y en las propiedades que las caracterizan;
a continuación, veremos cómo varían
dentro de la serie: no memorizaremos
estos hechos simplemente, sino que trataremos de comprender, hasta donde sea
posible, las propiedades en función de la estructura y las variaciones de las
propiedades en función de las variantes de estructura.
Después de haber estudiado con detalle el metano, analizaremos
ahora los miembros más complejos de la serie
de los alcanos. Estos hidrocarburos se han asignado a la misma familia
del metano por sus estructuras y, en
general, sus propiedades siguen la misma pauta establecida por el metano. Sin
embargo, surgirán algunas características nuevas, las cuales se deben
simplemente a su mayor tamaño y complejidad.
3.2 Estructura del etano
El que sigue en tamaño al metano es el etano, C2H6. Si
conectamos los átomos de esta molécula por enlaces covalentes, siguiendo la
regla de un enlace (un par de electrones) por cada hidrógeno y cuatro por cada
carbono (cuatro pares de electrones),
llegamos a la estructura
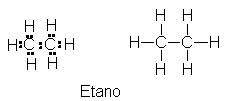
Cada carbono está
unido a tres hidrógenos y al otro carbono.
Puesto que cada átomo de carbono está unido a otros
cuatro átomos, sus orbitales enlazan tez (orbitales sp3) se dirigen
hacia los vértices de un tetraedro. Como en el caso del metano, los enlaces
carbono-hidrógeno resultan del solapamiento de estos orbitales sp3
con los s de los hidrógenos. La unión carbono-carbono surge del solapamiento de
dos orbitales sp3.
Los enlaces carbono-hidrógeno tienen la misma
distribución electrónica general, que es cilíndricamente simétrica en torno a
la línea de unión de los núcleos atómicos (véase Figura
3.1); debido a la similitud de sus formas, estos enlaces reciben el mismo
nombre: enlaces o (enlaces sigma.
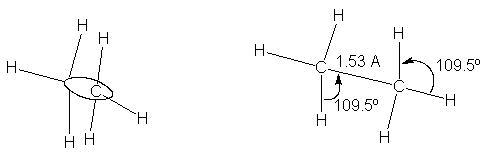
Fig. 3.1 Molécula de etano. Enlace simple Fig. 3.2 Molécula de etano: forma y dimensiones.
carbono-carbono: enlace o.
Así, los ángulos de enlace y las longitudes del enlace
carbono-hidrógeno del etano debieran ser muy similares a los del metano, es
decir, alrededor de 109.5º y 1.10 A, respectivamente, estructura que ha sido
confirmada en todos sus aspectos por difracción electrónica y estudios
espectroscópicos, obteniéndose para la molécula las medidas siguientes
electrónica y estudios espectroscópicos, obteniéndose para la molécula las
medidas siguientes (Fig.
3.2): ángulo de enlace, 109.5º; longitud C-H, 1.10 A; Longitud C-C, 1.53 A.
Estudios similares ha demostrado que
estos valores son, con variaciones sólo
muy ligeras, característicos de los
enlaces carbono-hidrógeno y carbono-carbono, y de los ángulos de enlace de los
alcanos.
3.3 Libre rotación
en torno al enlace simple carbono-carbono. Conformaciones. Tensión torsional
Este conjunto particular de ángulos y longitudes de
enlace no nos limita a un solo arreglo atómico para la molécula del etano,
puesto que no se especifica la relación entre los hidrógenos de un átomo de
carbono y los del otro. Al examinar los modelos para el etano (Fig. 3.3),
observamos que podríamos tener un ordenamiento como en I, en el que los
hidrógenos se hallan exactamente
opuestos, a un ordenamiento como en II, con los hidrógenos perfectamente
escalonados, o una infinidad de ordenamientos intermedios. ¿Cuál de éstos es la
verdadera estructura del etano? La respuesta es: todos ellos.
Hemos visto que el enlace o que une los átomos de carbono
es cilíndricamente simétrico en torno a la línea de unión de ambos núcleos de
carbono; los solapamientos y, consecuentemente, las energías de enlace deberían
ser iguales para todas estar ordenaciones posibles. Si las diversas
disposiciones no difieren en energía, entonces la molécula no queda restringida
a ninguna de ellas, sino que puede cambiar libremente de una a otra. Debido a
que tal cambio implica rotación en torno al enlace carbono-carbono. Describimos
esta libertad de cambio como que hay
libre rotación en torno al enlace simple carbono-carbono.
Los
ordenamientos atómicos diferentes que pueden intercambiarse por rotación en
torno a enlaces simples se denominan conformaciones. La I es la conformación
eclipsada, y la II, la conformación
escalonada. (La infinidad de conformaciones intermediarias se llaman sesgadas.)
Para representar las diferentes conformaciones, con
frecuencia utilizaremos dos tipos de fórmulas tridimensionales: fórmulas de caballete (Fig. 3.4);
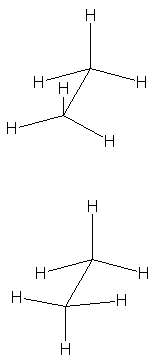
Fig. 3.4 Fórmulas de caballete para el etano en las conformaciones eclipsada y escalonada.
Y las denominadas proyecciones
de Newman, por M. S. Newman (Universidad del Estado de Ohio), quien propuso
por primera vez su uso.
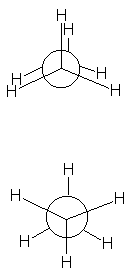
Fig.
3.5 Proyecciones de Newman para el etano, en las
conformaciones eclipsada y escalonada.
El cuadro aún no es completo; ciertas propiedades físicas indican que la rotación no es enteramente libre: hay una barrera
energética de unas 3 kcal/mol. La energía potencial de la molécula es mínima
para la conformación escalonada, aumenta con la rotación, y alcanza un máximo
para la conformación eclipsada (Fig. 3.6). La mayoría de las moléculas de etano
existen, naturalmente, en la conformación más estable, la escalonada; o bien, formulándolo de otro modo, toda
molécula pasa la mayor parte del tiempo en la conformación más estable.
¿Qué libertad tienen las moléculas de etano para rotar de
una disposición escalonada a otra? La barrera de 3 Kcal. No es muy alta: aun a
temperatura ambiente la fracción de colisiones con energía suficiente es
bastante grande, de modo que la
interconversión de ordenamientos escalonados es rápida. Con fines prácticos,
podemos seguir considerando que el enlace simple carbono-carbono permite la
rotación libre.
La naturaleza de la barrera rotacional se desconoce o lo
que no exactamente lo mismo no permite una explicación simple: Es demasiado
alta para que obedezca únicamente a las
fuerzas de Van der Waals (Sec. 1.19); a pesar de que los hidrógenos de carbonos opuestos son forzados a juntarse
más en la conformación eclipsada que en la escalonada, no son
suficientemente voluminosos como para que se produzca aglomeración apreciable,
véase Fig. 3.3. Se considera que la barrera surge en alguna forma de la interacción entre las nubes electrónicas de los enlaces carbono-hidrógeno. Cálculos de mecánica cuántica demuestran que la barrera debe
existir, de modo que quizá la <<falta
de comprensión>> sólo
sea una dificultad para parafrasear las matemáticas en términos físicos. Al
igual que los orbitales de enlace del metano, los dos juegos de orbitales del
etano tienden a apartarse lo más que puedan, a escalonarse.
La energía requerida para rotar la molécula de etano en
torno al enlace carbono-carbono se llama energía
torsional. Decimos que la inestabilidad relativa de la conformación
eclipsada o de cualquiera de las sesgadas intermediarias se debe a la tensión torsional.
A medida que se reemplazan los hidrógenos del etano por otros átomos o grupos de ellos,
aparecen otros factores que afectan a la estabilidad relativa de las
conformaciones: fuerzas de Van der Waals, interacciones dipolo-dipolo, puentes
de hidrógeno. No obstante, permanece la tendencia de los orbitales de enlaces
sobre carbonos adyacentes de localizarse escalonadamente, de modo que toda
rotación que se aleje de esa conformación va acompañada de tensión torsional.
Parte 4
3.4 Propano y
butanos
El miembro siguiente de la serie de los alcanos es el propano, C3H8.
Siguiendo una vez más la regla de un enlace por hidrógeno y cuatro por carbono,
llegamos a la estructura I.
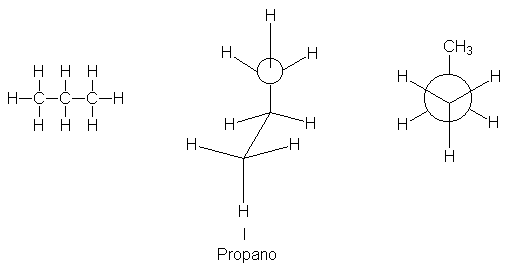
Aquí, la rotación puede producirse en torno a dos enlaces
carbono-carbono, y de nuevo es esencialmente libre. A pesar de que el grupo
metilo es considerablemente más grande que un hidrógeno, la barrera rotacional
es sólo ligeramente mayor (3.3 kcal/mol) que para el etano. Sin duda, la
aglomeración en la conformación eclipsada aún no es significativa, por lo que
la barrera rotacional se debe esencialmente al mismo factor que la barrera del
etano:
tensión
torsional (véase Fig. 3.7).
Cuando consideremos el butano, C4H10, nos encontramos con dos
estructuras posibles, II y III. La estructura II tiene una cadena de cuatro
carbonos, y la III, una de tres, con una ramificación de un carbono. No cabe
duda de que éstas representan dos estructuras diferentes, puesto que ningún
movimiento, torsión o rotación en torno a enlaces carbono-carbono permite
hacerlas coincidir; podemos observar
que en la estructura de cadena recta (II), cada carbono tiene por lo
menos dos hidrógenos, mientras que en
la de cadena ramificada (III) uno de los carbonos tiene solamente un
hidrógeno, o podemos apreciar que en el III un carbono está unido a otros
tres, mientras que en la II ningún carbono se enlaza con más de dos.
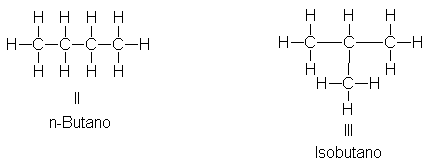
De acuerdo con esta predicción, nos encontramos con que
se conocen dos compuestos de la misma fórmula C4H10. Es
indudable que se trata de dos sustancias diferentes, puesto que presentan
diferencias apreciables en sus
propiedades físicas y químicas (véase Tabla 3.1); por ejemplo, uno hierve a 0ºC, y el otro, a
-12ºC; por definición, son isómeros
(Sec. 1.23).
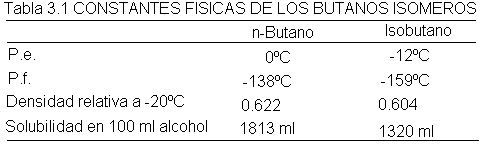
Se conocen dos compuestos de fórmula C4H10
y hemos dibujado dos estructuras para representarlos. La pregunta
siguiente es: ¿Qué estructura representa a uno y otro? Para responder, nos
valemos de la prueba del número de
isómeros. Al igual que el metano, los butanos pueden clorarse, reacción que puede llevarse hasta la
introducción de dos átomos de cloro por molécula. Del butano de p.e. 0ºC se
obtienen seis productos isómeros de
fórmula C4H8CI2, mientras que del de p.e.
-12ºC, solamente tres. Podemos dibujar justamente seis diclorobutanos con
cadena recta y solamente tres con cadena ramificada, por lo que el butano de
p.e. 0ºC debe ser de cadena recta, y el de p.e. -12ºC, ramificado. Para
distinguir ambos isómeros, la estructura recta se denomina n-butano (leído butano
normal), y la ramificada, isobutano.
3.5 Conformaciones del n-butano. Repulsión de Van der Waals
Observaciones más detenidamente la molécula del n-butano
y sus conformaciones. Fijando nuestra atención en el enlace C-C central, vemos
una molécula similar al etano, pero con un grupo metilo reemplazando dos
hidrógenos en cada carbono. Al igual que para el etano, las conformaciones escalonadas tienen energías torsionales menores, y son más estables que las eclipsadas. Sin embargo, debido a la
presencia de los grupos metilo, se encuentran aquí dos aspectos nuevos: en primer lugar, hay varias conformaciones
escalonadas diferentes y, además, las estabilidades conformacionales
se ven afectadas por un factor adicional al de la tensión torsional.
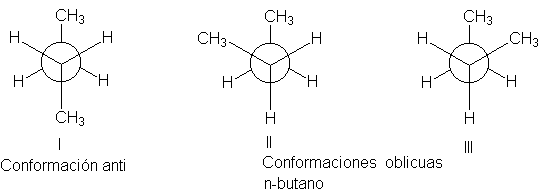
Existe la conformación anti, I, donde los grupos metilo se encuentran apartados al máximo
(ángulo diedro 180ºC), y hay dos conformaciones oblicuas, II y III, donde los metilos están separados sólo 60º.
(Las conformaciones II y III son imágenes especulares entre sí y tienen la
misma estabilidad; a pesar de esto, son diferentes. Construya modelos y
convénzase de que es así.)
Se ha determinado que la conformación anti es más estable (en 0.8 kcal/mol)
que la oblicua (Fig. 3.8). Ambas están libres de tensión
torsional, pero en la oblicua los metilos se aglomeran; es decir, se encuentran
a una distancia menor entre sí que la suma de sus radios de Van der Waals; en
estas circunstancias, las fuerzas de Van der Waals son repulsivas (Sec. 1.19),
por lo que elevan la energía de la conformación. Se habla de repulsión de Van der Waals (o repulsión estérica) entre los grupos
metilo, y de que la molécula es menos
estable, debido a la tensión de Van der
Waals (o tensión estérica). La aglomeración se observa claramente
utilizando modelos a escala (Fig. 3.9).
La tensión de Van der Waals puede afectar no sólo las
estabilidades relativas de las diversas conformaciones escalonadas, sino
también las alturas de las barreras entre ellas. El máximo energético que se
alcanza cuando ambos metilos se cruzan es
la barrera más alta aún mayor que cuando se cruzan un metilo y un
hidrógeno - y ha sido estimada en 4.4-6.1 kcal/mol; aun así, es suficientemente baja como para que
la energía de las colisiones moleculares provoque rotación rápida - al menos a
temperaturas ordinarias -, de modo que, en un instante, existe una molécula
determinada en una conformación oblicua,
y en el próximo, en la anti.
Volveremos más adelante sobre las relaciones entre
conformaciones del n-butano como éstas.
(Sec. 4.20).
3.6 Alcanos
superiores. La serie homóloga
Si examinamos las fórmulas moleculares de los alcanos considerados hasta ahora, observamos
que el butano contiene un carbono y dos hidrógenos más que el propano, que, a su vez, tiene un carbono y dos
hidrógenos más que el etano, y así
sucesivamente. Una serie de compuestos
cuyos miembros difieren del siguiente en un valor constante se denomina serie
homóloga, y sus miembros son homólogos.
La familia de los alcanos forma tal
serie homóloga, siendo la diferencia
constante entre miembros
sucesivos igual a CH2. También apreciamos que en cada uno de estos alcanos, el número de hidrógenos es más
de dos veces el doble del número de
átomos de carbono, por lo que podemos escribir una fórmula general para sus
miembros, que es CnH2n+2. Como veremos más
adelante, otras series homólogas tiene sus propias fórmulas generales
características.
De acuerdo con
esta fórmula general, el próximo alcano, el
pentano, corresponde a C5H12, seguido del hexano, C6H14, el heptano, C7H16, y
así sucesivamente. Es de esperar que, a medida que crezca el número de átomos,
también aumente el número de ordenamientos posibles, lo que efectivamente
resulta ser el caso; el número de isómeros de homólogos sucesivos
aumenta de modo sorprendente: hay 3 pentanos isómeros, 5 hexanos, 9
heptanos y 75 decanos (C10);
para el eicosano de 20 carbonos hay 366 319
estructuras isómeras posibles.
Los esqueletos carbonados de los pentanos y hexanos
isómeros se indican a continuación:
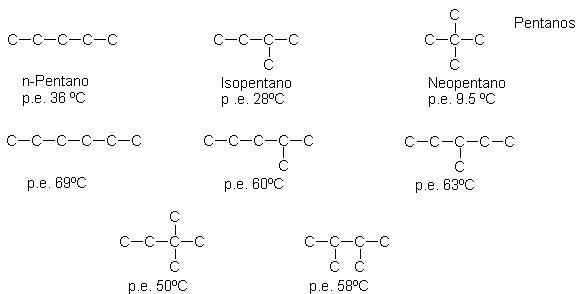
Es importante practicar el dibujo de las estructuras isómeras posibles que corresponden a una fórmula molecular única. Al hacer esto, es particularmente útil contar con un juego de modelos moleculares, porque con él se puede detectar que muchas estructuras que parecen diferentes al dibujarlas sobre el papel, son realmente idénticas.
3.7 Nomenclatura
Vimos que se emplean los nombres metano, etano, propano, butano y pentano para alcanos con uno, dos,
tres, cuatro y cinco átomos de carbono, respectivamente. La tabla 3.2 indica
los nombres de muchos alcanos superiores. Excepto para los cuatro primeros
miembros de la familia,

el nombre simplemente
se deriva del prefijo griego (o
latino) para el número particular de
carbonos en el alcano, de modo que resulta pentano
para cinco, hexano para seis, heptano para siete, octano para ocho,
etc.
Deben memorizar los nombres de por lo menos los diez primeros alcanos. Logrado esto, se habrán
aprendido simultáneamente también, en esencia, los nombres de los diez primeros alquenos, alquinos, alcoholes, etc, puesto que los nombres de
muchas familias de sustancias están íntimamente relacionados; por ejemplo,
compárense los nombres propano,
propeno y propino, para el alcano, alqueno y alquino de tres
carbonos.
Sin embargo, prácticamente todo alcano puede tener cierto
número de estructuras isómeras, debiendo haber un nombre inequívoco para cada una de ellas. Los
butanos y pentanos se distinguen por el empleo de prefijos: n-butano e isobutano; n-pentano, isopentano y neopentano. Pero hay 5 hexanos, 9 heptanos y 75 decanos; sería
difícil encontrar y, aún más, recordar
un prefijo para cada uno de estos isómeros; es evidente que se necesita algún
método sistemático para nombrarlos.
Durante el desarrollo de la química orgánica, se han
inventado diferentes métodos para nombrar los miembros de prácticamente todos
los tipos de compuestos orgánicos; cada
método se ideó una vez que el sistema
empleado antes resultaba inadecuado para el creciente número de sustancias orgánicas cada vez más
complejas. Es, quizá, lamentable para
nosotros que hayan sobrevivido varios sistemas y que aún sean de uso corriente.
Aun cuando nos contentemos con emplear un solo sistema, es necesario entender
los nombres usados por otros químicos,
por lo que debemos aprender más de un sistema de nomenclatura; pero antes de
emprender la tarea, debemos conocer previamente los nombres de ciertos grupos
orgánicos.
3.8 Grupos
alquilo
Durante el estudio de la química inorgánica, se vio la
utilidad de tener nombres para ciertos grupos de átomos que sólo constituyen
parte de una molécula y que sin embargo aparecen muchas veces como una unidad:
por ejemplo, NH4+, amoniaco;
NO3- nitratos;
SO32-, sulfito;
etcétera.
De modo similar, se dan nombres a ciertos grupos que
aparecen constantemente como unidades
estructurales de moléculas orgánicas. Vimos que el clorometano, CH3CI,
también se conoce como cloruro de metilo.
El grupo CH3 se llama metilo
donde aparezca, por lo que CH3Br es bromuro de metilo;
CH3I, yoduro de metilo; CH3OH,
alcohol metílico. De forma similar,
el grupo C2H5 se llama etilo; C3H7, propilo; C4H9, butilo; etc.
Estos grupos se
nombran cambiando simplemente la terminación ano del alcano
correspondiente por ilo. Colectivamente, se les conoce como grupos alquilo. La fórmula general para
un grupo alquilo es CnH2n+1, ya que contiene un hidrógeno
menos que el alcano correspondiente, CnH2n+2.
Otra vez encontramos el problema de isomería entre los
grupos alquilo. Sólo existe un cloruro de metilo o de etilo y, correspondientemente,
un solo grupo metilo o etilo. Sin embargo, podemos apreciar que hay dos
cloruros de propilo, I y II, por lo que deben existir dos grupos propilo. Ambos contienen la cadena propano, pero
difieren en el punto de unión
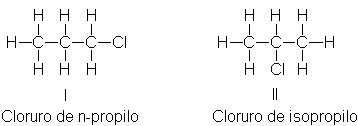
Con el cloro; se denominan n-propilo e isopropilo, Podemos distinguir los dos cloruros por los nombres cloruro de n-propilo y de isopropilo,
respectivamente; de la misma forma se hace con los dos bromuros, yoduros,
alcoholes, etc., correspondientes

Hay cuatro grupos butilo, dos que derivan del n-butano de
cadena recta, y dos del isobutano ramificado: se les designa n-(normal),
sec-(secundario), iso- y t-
(terciario), como se ilustra a continuación. Nuevamente, la diferencia se encuentra en el punto de unión
del grupo alquilo con el resto de la molécula.
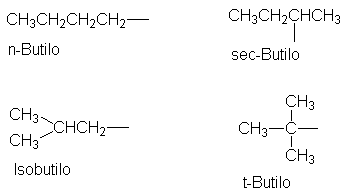
Más allá del butilo, el número de grupos isómeros
derivados de cada alcano se hace tan
grande que resulta impráctico designarlos todos con prefijos. Aunque limitado,
este sistema es tan útil para los grupos pequeños recién descritos, que se
emplea con frecuencia; el estudiante
debe, por tanto, memorizar estos nombres y aprender a reconocer los grupos a primera vista, cualquiera que sea la forma en que aparezcan representados.
Por muy grande que sea un grupo, una de sus muchas
disposiciones posibles puede ser
designada siempre por este sistema. El prefijo n- siempre se emplea para
indicar un grupo alquilo cuyos carbonos forman una sola cadena continua y cuyo
punto de enlace es el carbono del extremo; por ejemplo:

El prefijo iso se usa para designar cualquier grupo
alquilo (de seis carbonos o menos) que tiene una sola ramificación
monocarbonada en el penúltimo carbono de la cadena y con el punto de unión en
el extremo opuesto de ella. Por ejemplo:
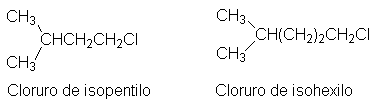
Si la raificación
aparece en otra posición cualquiera, o si el punto de unión está en otra
posición, este prefijo no es aplicable.
Ahora que hemos aprendido los nombres de algunos grupos alquilo, volvamos al problema
original: la designación de los
alcanos.
3.9 Nombres
comunes de los alcanos
Hemos visto que los prefijos n-, iso- y neo- son adecuados para diferenciar los diversos butanos
y pentanos, pero más allá de éstos se necesitaría un número irracional de
prefijos. Sin embargo, se retiene el prefijo n- para cualquier alcano, por
largo que sea, en el cual todos los carbonos forman una cadena continua sin
ramificaciones:

Un isoalcano es
un compuesto de seis carbonos o menos en el que todos ellos, excepto uno,
forman una cadena continua, con el carbono no considerado unido al penúltimo:
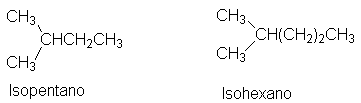
Para designar cualquiera de los alcanos superiores usamos
el sistema IUPAC, presentado en la sección siguiente.
(A veces, es conveniente denominar los alcanos como
derivados del metano; véase, por ejemplo, I en Sec. 4.13.)
3.10 Nombres
IUPAC de alcanos
Para diseñar un sistema de nomenclatura aplicable a los
compuestos aún más complejos, se han reunido periódicamente desde 1892 varios
comités y comisiones en representación de los químicos del mundo. En su forma
actual, el sistema diseñado se conoce como
sistema IUPAC (International Union of Pure and Applied
Chemistry); dado que sigue, más o menos, la misma pauta para todas las
familias de compuestos orgánicos, lo consideraremos en detalle para su
aplicación a los alcanos.
En esencia, las reglas del sistema IUPAC son:
1. Como estructura de referencia, seleccione la cadena continua más larga y considere luego que el compuesto deriva de aquella, reemplazando los hidrógenos por diversos grupos alquilo. Así, puede considerarse que el isobutano (I) deriva del propano por reemplazo de un átomo de hidrógeno por un grupo metilo, por lo que se puede denominar metilpropano.
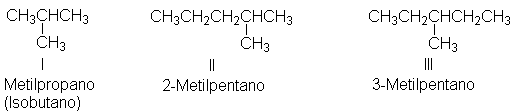
2. Donde sea necesario, como es el caso de los dos metilpentanos isómeros (II y III), indique el carbono Que lleva el grupo alquilo por medio de un número.
3. Al numerar la cadena carbonada de referencia, comience
por el extremo que resulte en el empleo de Los números más bajos; así, II se denomina 2-metilpentano,
y no 4-metilpentano.
4.
Si un mismo grupo alquilo aparece más de una vez como
cadena lateral, indíquelo por los prefijos di-, tri-, tetra-, etc., para destacar
cuántos de esos alquilos hay, e indique con números la posición de cada grupo,
como en 2,2,4-trimetilpentano (IV).
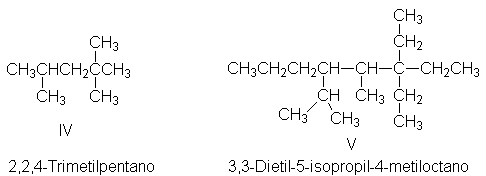
5. Si a la cadena principal se unen varios grupos alquilo
diferentes, nómbrelos en orden alfabético, como en 3,3-dietil-5-isopropil-4-metiloctano (V). (Obsérvese que isopropil aparece antes que metil; sin embargo, dimetil debe aparecer después que etil
o dietil.)
Hay reglas y
convenciones adicionales que se emplean para nombrar alcanos muy complicados,
pero las cinco reglas fundamentales mencionadas aquí son suficientes para los
compuestos que encontraremos.
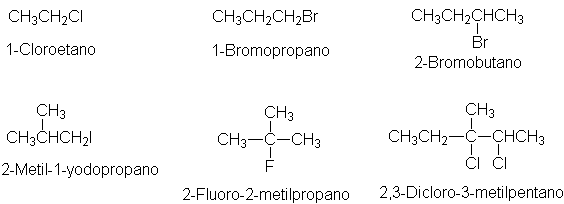
Los halogenuros de alquilo, que aparecen muchas veces en
la química de los alcanos, se denominan haloalcanos;
es decir, se trata el halógeno como si fuese sólo una cadena lateral. Nombramos
primero el alcano como si no contuviese halógeno, y agregamos a continuación fluoro, cloro, bromo o yodo, junto con
los números y prefijos que fuesen necesarios.
3.11 Tipos de
átomos de carbono e hidrógeno
Se ha encontrado que es de gran utilidad clasificar cada átomo de carbono de un alcano de acuerdo con el número de átomos de carbono adicionales que tiene unidos. Un átomo de carbono primario (1º) está unido a un solo carbono adicional; uno secundarios (2º), a otros dos; y uno terciario (3º), a tres. Por ejemplo:
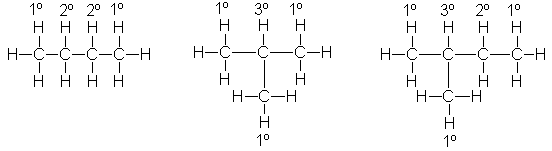
Cada átomo de hidrógeno se clasifica de forma similar,
recibiendo la misma designación de primario,
secundario o terciario, según el carbono al cual se encuentre unido.
Al considerar las reactividades relativas de las
diferentes partes de una molécula de alcano, haremos uso constante de estos
nombres.
3.12 Propiedades
físicas
Las propiedades físicas de los alcanos siguen el mismo
patrón establecido por el metano, siendo concordantes con las estructuras de
los alcanos. La molécula de un alcano sólo presenta enlaces covalentes, que
enlazan dos átomos iguales, por lo que
no son polares; o bien, unen dos átomos
cuyas electronegatividades apenas difieren, por lo que son escasamente polares.
Además, estos enlaces son direccionales de un modo muy simétrico, lo que
permite que dichas polaridades débiles se cancelen; como resultado, un alcano
es no polar o ligeramente polar.
Hemos visto (Sec. 1.19) que las fuerzas que mantienen
unidas las moléculas no polares (fuerzas de Van der Waals) son débiles y de
alcance muy limitado; solamente actúan entre partes de moléculas diferentes en
contacto íntimo; es decir, entre las superficies moleculares. Dentro de una
familia esperaríamos que cuanto mayor sea una molécula y por consiguiente su
superficie-, más intensa son las fuerzas intermoleculares.
La tabla 3.3. registra algunas constantes físicas para unos
pocos n-alcanos. Podemos apreciar que los puntos de ebullición y fusión
aumentan a medida que crece el número de
carbonos. Los procesos de ebullición y fusión requieren vencer las fuerzas intermoleculares de un líquido y
un sólido; los puntos de ebullición y fusión
suben porque dichas fuerzas se intensifican a medida que aumenta el
tamaño molecular.
Salvo para los alcanos muy pequeños, el punto de ebullición aumenta de 20 a 30 grados por cada carbono que
se agrega a la cadena; veremos que este incremento de 20 a 30 grados por
carbono no

Sólo se cumple para los alcanos, sino también para todas
las series homólogas que estudiaremos.
El aumento del punto de fusión no es tan regular, debido a
que en un cristal las fuerzas intermoleculares no sólo dependen del tamaño de
las moléculas, sino también de su ajuste en el retículo cristalino.
Los cuatro primeros n-alcanos son gases; como resultado
del aumento del punto de ebullición y
punto de fusión con la longitud creciente de la cadena, los trece siguientes (C5-C17)
son líquidos, y los de 18 átomos de
carbono o más, sólidos.
Las diferencias en los puntos de ebullición de los
alcanos con igual número de carbonos,
pero estructura distinta, son algo menores. En la tabla 3.1 y en la
sección 3.6 figuran los puntos de ebullición de los butanos, pentanos y hexanos
isómeros; se aprecia que, en cada caso, un
isómero ramificado tiene un punto de ebullición más bajo que uno de cadena
recta y, además, cuanto más numerosas son las ramificaciones, menor es el
punto de ebullición correspondiente. Así, el n-butano hierve a 0ºC, y el
isobutano, a -12ºC; el n-pentano tiene
un punto de ebullición de 36ºC;
el isopentano con una ramificación, 28ºC, y el
neopentano con dos, 9.5ºC. Este efecto sobre los puntos de ebullición de
las ramificaciones se observa en todas las familias de los compuestos
orgánicos. El hecho de que una ramificación baje los puntos de ebullición es
razonable: con la ramificación, la
forma de la molécula tiende a aproximarse a la de una esfera, con lo que
disminuye su superficie. Esto se traduce en un debilitamiento de las fuerzas
intermoleculares que pueden ser superadas a temperaturas más bajas (Sec. 1.20).
Compárense, por ejemplo, las formas de los pentanos isómeros que aparecen en la
figura 3.10.
De acuerdo con la regla empírica, <<una sustancia disuelve a
otra similar>>, los
alcanos son solubles en disolventes no polares como benceno, éter y cloroformo,
e insolubles en agua y otros disolventes fuertemente polares. Considerándolos
como disolventes, los alcanos líquidos
disuelven compuestos de polaridad baja, pero no los de alta.
La densidad de los alcanos aumenta en fusión del tamaño,
tiende a nivelarse en torno a 0.8, de
modo que todos ellos son menos densos que el agua. No es de sorprender que casi
todos los compuestos orgánicos sean
menos densos que el agua, puesto que, al igual que los alcanos, están constituidos principalmente
por carbono e hidrógeno. En general, para que
una sustancia sea más densa que el agua, debe contener un átomo pesado,
como el bromo o el yodo, o varios átomos, como el cloro.
3.13 Fuente
industrial
La fuente principal de alcanos es el petróleo, junto con el gas
natural que lo acompaña. La putrefacción y las tensiones geológicas han
transformado, en el transcurso de millones de años, compuestos orgánicos
complejos que alguna vez constituyeron
plantas o animales vivos en una
mezcla de alcanos de 1 hasta 30 ó 40 carbonos. Junto a ellos, y particularmente
abundantes en el petróleo de California, se encuentran los cicloalcanos (Cap. 12), que en la industria petrolera se conocen
como naftenos.
Una segunda fuente potencial de alcanos la constituye el
otro combustible fósil, el carbón; se están
desarrollando procesos que lo convierten, por medio de la hidrogenación,
en gasolina y petróleo combustible,
como también en gas sintético, para contrarrestar la escasez previsible del gas
natural.
Evidentemente, el gas natural sólo contiene los alcanos
más volátiles, es decir, los de bajo peso molecular; está constituido en
esencia por metano y cantidades progresivamente menores de etano, propano y
alcanos superiores. Una muestra obtenida de un oleoducto alimentado por gran número de pozos de
Pensilvania contenía metano, etano y propano
en proporción de 12:2:1; los
alcanos superiores representaban sólo el 3% del total. La fracción
propano-butano se separa de los
componentes más volátiles por licuación, se comprime en cilindros y se vende como
gas licuado en áreas que no tienen
gas de alumbrado.
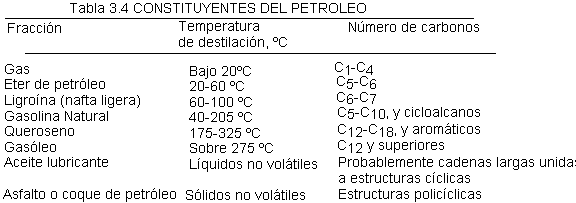
El petróleo se separa por destilación en las diversas
fracciones enumeradas en la tabla 3.4; debido a la relación entre punto de
ebullición y peso molecular, esto supone
una separación preliminar de acuerdo con el número de carbonos. Cada
fracción aún es una mezcla compleja, sin embargo, puesto que contiene alcanos
con un intervalo de átomos de carbono y cada número de carbonos representa
varios isómeros. El uso de cada fracción depende principalmente de su
volatilidad o viscosidad, e importa muy poco si es una mezcla compleja o un solo compuesto puro. (En Sec.
3.30 veremos que la estructura de los
componentes de la gasolina es de importancia fundamental.)
El principal uso de todas las fracciones volátiles es utilizarlo como combustibles. La
fracción gaseosa, igual que el gas natural, se emplea sobre todo en
calefacción. La gasolina se utiliza en máquinas de combustión interna que
requieren un combustible bastante
volátil; el queroseno se usa en
motores de tractor y reactores, y el
gasóleo, en motores Diesel. Estos dos últimos también se emplean para
calefacción, conociéndose también el último como fuel-oil.
La fracción de
aceite lubricante, especialmente la procedente de crudos de Pennsilvania (petróleo de base parafínica), a menudo
contiene grandes cantidades de alcanos de cadena larga (C20-C34),
con puntos de fusión bastante altos. Si éstos permanecieran en el aceite, en días fríos podrían cristalizar en forma
de sólidos cerosos en los oleoductos; para prevenirlo, se enfría el aceite y se
separa la cera por filtración, que se vende como cera parafínica (p.f. 50-55ºC) una vez purificada, o bien se emplea
como gelatina de petrolato
(vaselina). El asfalto se emplea para impermeabilizar techumbres y en la
pavimentación de carreteras. El coque obtenido de crudos de base parafínica se
compone de hidrocarburos complejos de
elevada proporción de carbono a hidrógeno; se usa como combustible o en la manufactura de electrodos
De carbono para la industria electroquímica. El éter del
petróleo y la ligroína son disolventes útiles para muchos materiales orgánicos
de baja polaridad.
Además de emplearse directamente como se acaba de describir,
ciertas fracciones del petróleo se convierten en otras clases de compuestos
químicos. La isomerización catalítica
transforma alcanos de cadena recta en
ramificados; el proceso cracking
(Sec. 3.31) convierte alcanos
superiores en inferiores y en alquenos, con lo que se aumenta el
rendimiento de la gasolina: incluso
puede usarse para la producción de <<gas
natural>>. Es
más, los alquenos así formados constituyen quizá las materias primas más importantes para la síntesis de sustancias alifáticas en gran escala. El proceso de reformación catalítica (Sec. 15.5) convierte los alcanos y los
cicloalcanos en hidrocarburos aromáticos, con lo que contribuye a proporcionar
materias primas para la síntesis en gran escala de otra amplia gama de compuestos.
3.14 Fuente
industrial y preparación de laboratorio
Generalmente dividiremos los métodos para obtener un tipo
particular de compuesto en dos categorías: fuente
industrial y preparación de laboratorio. Podemos comparar ambas como se
explica a continuación, aunque debe tenerse presente que hay muchas excepciones
a estas generalizaciones.
Una fuente industrial debe proporcionar grandes
cantidades del material deseado y al menor costo posible. En un laboratorio,
tal vez se necesite preparar unos cuantos cientos de gramos o incluso unos
pocos gramos: el costo suele tener
menor importancia que el tiempo del investigador.
Para muchos fines industriales puede ser igualmente
apropiada una mezcla que una sustancia pura; aun cuando se requiera un
compuesto único, puede resultar factible económicamente separarlo de una
mezcla, en particular si los demás componentes son comerciales. En el
laboratorio, en cambio, el químico casi siempre necesita una sola sustancia
pura: la separación de un compuesto de
una mezcla de materiales similares consume mucho tiempo, y a menudo no
proporciona un compuesto de la pureza requerida. Además, la materia prima para una preparación particular, bien puede
ser el producto obtenido laboriosamente de una síntesis previa o incluso de una
serie de preparaciones, por lo que conviene convertirlo lo más completamente posible en su compuesto deseado. A escala
industrial, si no es posible aislar una sustancia de un material de origen
natural, se puede sintetizar junto con varios compuestos similares por medio de
alguna reacción económica. Siempre que sea posible, en el laboratorio se elige
un proceso que forme un solo compuesto de alto rendimiento.
En la industria, a menudo es conveniente desarrollar un proceso y diseñar el equipo capaz de
sintetizar un solo miembro de una
familia química. En el laboratorio, raras veces un químico se interesa en
preparar el mismo compuesto una y otra vez, por lo que emplea métodos aplicables a muchos o a todos los
componentes de una familia específica.
En nuestro estudio de la química orgánica, concentraremos
más nuestra atención en las preparaciones versátiles de laboratorio que en los
limitados método industriales. Al
analizar aquellos, y por simplicidad,
emplearemos como ejemplos, la
preparación de compuestos que, de hecho
nunca se obtienen por el método
indicado. Podríamos estudiar la síntesis del etano por medio de la
hidrogenación del etileno, aunque estemos
en condiciones de adquirir de la
industria petrolera todo el etano que necesitamos. Sin embargo, si
sabemos cómo convertir etileno en
etano, también sabremos cómo transformar 2-metil-1-hexeno en 2-metilhexano
cuando lo necesitamos, o colesterol en
colesterol o, si quisiéramos, aceite de semilla de algodón en oleomargarina.
3.15 Preparación.
Cada uno de los alcanos menores, desde el metano hasta el
n-pentano y el isopentano, puede obtenerse en forma pura por destilación
fraccionada del petróleo y del gas natural;
el neopentano no existe en la
naturaleza. Más allá de los pentanos, el número de isómeros de cada homólogo se
hace tan grande y las diferencias en
sus puntos de ebullición tan pequeñas,
que resulta imposible aislar compuestos individuales puros; estos
alcanos deben ser sintetizados por alguno de los métodos expuestos más
adelante.
En algunas de las
reacciones se emplea el símbolo R para
representar cualquier grupo alquilo,
recurso conveniente para resumir reacciones
que son típicas de una familia entera y que enfatiza la similitud
general de sus miembros.
Sin embargo, ale escribir estas ecuaciones generales, no
debe perder de vista un aspecto importante; Para tomar un ejemplo específico,
una reacción que involucra RCI sólo tiene significado en función de una
reacción que podamos realizar en el laboratorio empleando un compuesto
real, como el cloruro de metilo o de
t-butilo. Aunque típica de los
halogenuros de alquilo, una reacción puede variar ampliamente en
velocidad o rendimiento, lo que depende
del grupo alquilo específico implicado. Probablemente, debamos emplear
condiciones experimentales muy diferentes para el cloruro de metilo que para el
de t-butilo; en un caso extremo, un proceso que funciona bien para el cloruro
de metilo, puede ser tan lento o dar tantos subproductos que resulta
completamente inútil para el cloruro de t-butilo.

De los métodos que se verán más adelante, la
hidrogenación de alquenos es, con mucha diferencia, el más importante. Al
agitar bajo una ligera presión de hidrógeno en presencia de una pequeña
cantidad de un catalizador, los alquenos se convierten suave y
cuantitativamente en alcanos con el
mismo esqueleto carbonado. La única limitación del procedimiento es la asequibilidad
del alqueno apropiado, la cual no es muy seria, pues los alquenos (Sección
7.11) se pueden preparar fácilmente de
alcoholes, que, a su vez, pueden sintetizarse sin dificultad (Sec. 17.8) en
gran variedad de tamaños y formas.
La reducción de un halogenuro de alquilo, ya por medio de
un reactivo de Grignard, ya directamente con metal y ácido, implica sólo el
reemplazo de un átomo de halógeno por uno de hidrógeno; el esqueleto carbonado
permanece intacto. Este método tiene
casi la misma aplicabilidad que el
anterior, puesto que los halogenuros de alquilo, al igual que los alquenos, generalmente se preparan a partir de alcoholes. En los casos en que
pudieran usarse ambos procedimientos,
quizá sea preferible la hidrogenación de los alquenos, debido a su mayor sencillez y mayor rendimiento.
El acoplamiento de los halogenuros de alquilo con
compuestos órgano metálico es el único de estos métodos que forma enlaces
carbono-carbono, generando un esqueleto carbonado nuevo y de mayor tamaño.
3.16 El
reactivo de Grignard: un compuesto organometálico
Cuando se pone en contacto una solución de un halogenuro de alquilo con éter etílico
seco, (C2H5)2O, con virutas de magnesio
metálico se produce una reacción vigorosa: la solución se hace lechosa, comienza
a hervir y el magnesio metálico desaparece gradualmente. La solución resultante
se conoce como reactivo de Grignard.
(Victor Grignard, de la Universidad de Lyons, obtuvo el Premio Nobel en 1912
por su descubrimiento.) Es uno de los reactivos más útiles y versátiles para el
químico orgánico.
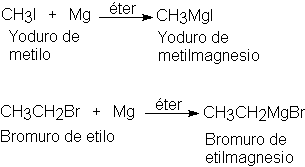
El reactivo de Grignard tiene la fórmula general RMgX, y
su nombre general es halogenuro de alquilmagnesio. El enlace carbono-magnesio
es covalente, pero muy polar: el carbono atrae electrones del magnesio electropositivo;
el enlace magnesio-halógeno es esencialmente iónico.
![]()
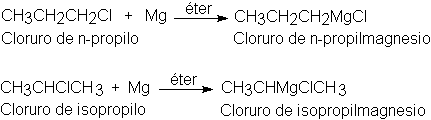
Puesto que el magnesio se une al mismo carbono que antes
tenía halógeno, el grupo alquilo permanece intacto durante la preparación del
reactivo; así, el cloruro de n-propilo da
cloruro de n-propilmagnesio, y el isopropilo se convierte en cloruro de
isopropilmagnesio.
El reactivo de Grignard es el miembro mejor conocido de
un tipo de sustancias, llamadas compuestos órgano
metálicos, que se caracterizan por la unión de carbono a un metal: litio,
potasio, sodio, cinc, mercurio, plomo, talio, y a casi todo metal conocido.
Cada tipo de compuesto órgano metálico, por supuesto, tiene su propio conjunto
de propiedades y sus usos específicos dependen
de ellas. Cualquiera que sea el metal, es menos electronegativo que el carbono, por lo que el enlace
carbono-metal como el del reactivo de Grignard es altamente polar. Aunque el
grupo orgánico no es un carbanión acabado
un anión que posee carbono con carga negativa (Sec. 7.18)-, tiene, sin embargo,
considerable carácter carbaniónico. Veremos
que los compuestos
organometálicos deben su enorme utilidad esencialmente a una propiedad común: pueden servir de fuente para
la transferencia de carbono con sus
electrones.
![]()
El compuesto de Grignard es muy reactivo: se combina con
numerosas sustancias inorgánicas que incluyen agua, dióxido de carbono y
oxígeno, y con la mayoría de los compuestos orgánicos; en muchos de estos
casos, la reacción proporciona la mejor ruta para obtener un tipo particular de sustancia orgánica.
La reacción con agua para formar un alcano es típica del comportamiento del reactivo de Grignard con los ácidos, y de muchas de las sustancias organometálicas más reactivas. En vista del marcado carácter carbaniónico del grupo alquilo, podemos considerar que el reactivo de Grignard es la sal magnésica, RMgX, del muy débil ácido R-H. La reacción
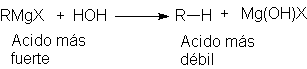
Es tan sólo el desplazamiento del ácido más débil, R-H,
de su sal por el ácido más fuerte, HOH.
Un alcano es un ácido tan débil que es desplazado del
reactivo de Grignard por compuestos que, de ordinario, consideraríamos como
ácidos muy débiles o ni siguiera como tales. Un compuesto con hidrógeno unido a
oxígeno o nitrógeno es mucho más ácido que un alcano, por lo que puede
descomponer un reactivo de Grignard: por ejemplo, amoniaco o
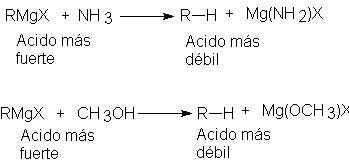
alcohol
metílico. Para la preparación de un alcano, un ácido es tan bueno como otro,
por lo que elegimos naturalmente el agua, que es el más accesible y cómodo.
3.17 Acoplamiento
de halogenuros de alquilo con compuestos organometálicos
Para hace un alcano de mayor número de átomos de carbono que el material de partida, se
requiere de la formación de enlaces carbono-carbono, siendo la manera más
directa de lograrlo el acoplamiento de dos grupos alquilo. El método más
versátil lo constituye la síntesis desarrollada en las postrimerías de la
década de 1960 por E. J. Corey y Herbert House, que trabajaban
independientemente en la Universidad de Harvard y en el Instituto Tecnológico
de Massachusetts, respectivamente. El acoplamiento se produce en la reacción
entre un cuprodialquil-litio, R2CuLi,
y un halogenuro de alquilo, R’X (R’ significa un grupo alquilo que puede ser
igual o diferente de R.)
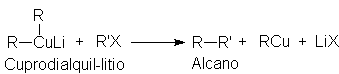
Se prepara un alquil-litio, Rli, a partir de un
halogenuro de alquilo, RX, del mismo modo que un reactivo de Grignard, al cual
se agrega un halogenuro cuproso, CuX, y finalmente, un segundo halogenuro de
alquilo, R’X. Por último, el alcano se sintetiza a partir de dos halogenuros de
alquilo: RX y R’X.
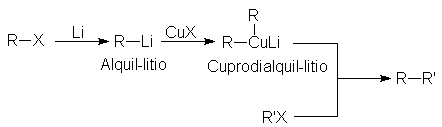
Para obtener buenos rendimientos, el R’X debe ser un
halogenuro primario; el grupo R del
Organometálico puede ser primario, secundario o terciario, como por ejemplo:
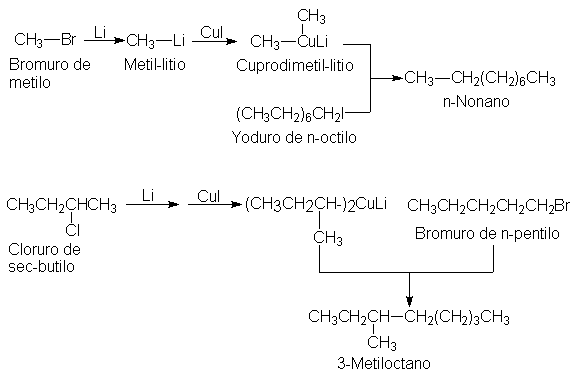
La elección
del reactivo organometálico es crucial: los reactivos de Grignard o los
alquil-litios, por ejemplo, sólo se acoplan con unos pocos halogenuros
orgánicos excepcionalmente reactivos;
los compuestos organosódicos se acoplan, pero son tan reactivos que se
unen durante su formación con su halogenuro de alquilo de origen. La reacción
del sodio con halogenuros de alquilo (reacción
de Wurtz) queda así limitada a la síntesis de alcanos simétricos. R-R.
Se sabe desde hace tiempo que los compuestos organocuprosos
son particularmente buenos para formar enlaces
carbono-carbono, pero son inestables. Aquí, son generados in situ partiendo del organolitio, combinándose luego con más de
éste para formar estos compuestos organometálicos relativamente estables, que existen
como agregados complejos, pero se cree
que corresponden aproximadamente a R2Cu-Li+. Este anión es un ejemplo de un
compuesto ato, la contrapartida
negativa de un complejo onio (amonio,
oxonio).
Aunque no se conoce bien el mecanismo, hay pruebas que
sugieren, al menos, lo siguiente: el grupo alquilo R es transferido desde el
cobre, con un par de electrones, y se une al alquilo R’ en lugar de un Ion
halogenuro (sustitución nucleofilica
alifática, Sec. 5.8).
3.18 Reacciones
A veces nos referimos a lso alcanos con el nombre
anticuado de parafinas. Este nombre (latín: parum
affinis, <<sin
afinidad suficiente>>) se
les dio para describir lo que parecía una reactividad baja de estos
hidrocarburos.
Sin embargo, la reactividad depende de la elección del
reactivo. Cuando los alcanos son inertes a los ácidos clorhídrico y sulfúrico,
reaccionan fácilmente con ácidos como son inertes a los ácidos como HF-SbF5
y FSO3H-SbF5 (<<ácido mágico>>) para dar varios
productos. A pesar de ser inertes hacia agentes oxidantes, como el permanganato
de potasio o el dicromato de sodio,
dedicaremos la mayor parte de este capítulo a su oxidación por
halógenos. Ciertas levaduras se alimentan perfectamente de alcanos para
producir proteínas, lo que, sin duda, es una reacción química. Como solía decir
el profesor M.S. Kharash (Sec. 8.6): <<Considérese
la “inercia” de una habitación llena de
gas natural, aire y un fósforo encendido>>
Aún así, comparativamente, su reactividad es limitada. El
<<ácido
mágico>> es,
después de todo, uno de los ácidos más fuertes que se conocen; la hidrogenación
requiere luz o calor, y la combustión precisa una llama o chispa para
iniciarse.
Una parte importante de la química de lso alcanos implica
reacciones de radicales libres, que tienen lugar en condiciones vigorosas y dan
generalmente mezclas de productos. Se necesita una partícula reactiva
típicamente, un átomo o un radical libre para iniciar el ataque a una molécula
de alcano. Es la generación de esta partícula reactiva lo que necesita las
condiciones vigorosas: por
ejemplo, la disociación de una molécula
de halógeno en átomos o incluso la disociación de la propia molécula de alcano
(como en la pirolisis).
Durante su ataque, la partícula reactiva le quita
hidrógeno al alcano, con lo que éste se transforma en una partícula reactiva
que continúa la secuencia de la reacción; es decir, mantiene la cadena. Sin embargo, la molécula del alcano contiene
muchos átomos de hidrógeno, por lo que el producto específico que se obtenga
dependerá de cuál de ellos se extrae. Aunque una partícula atacante puede
exhibir cierta selectividad, puede extraer un hidrógeno de cualquier parte de
la molécula, provocando la formación de muchos productos isómeros.
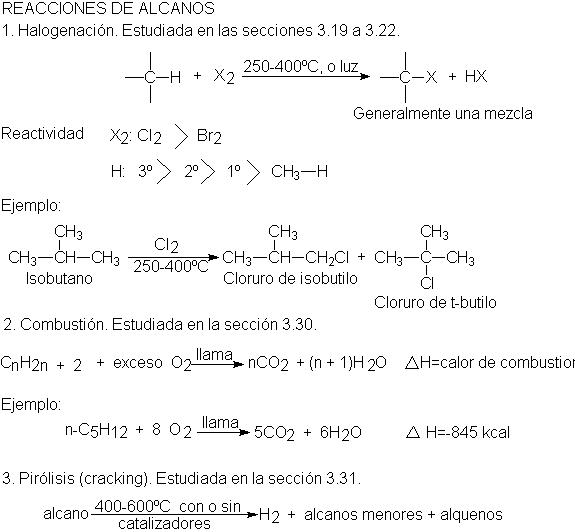
3.19 Halogenación
Como es de esperar, la halogenación de los alcanos
superiores es esencialmente igual a la del metano. Sin embargo, puede
complicarse por formación de mezclas de isómeros.
Bajo la influencia de la luz ultravioleta, o a 250-400ºC,
el cloro o bromo convierte los alcanos en cloroalcanos (cloruros de alquilo) o
bromoalcanos (bromuros de alquilo), formándose simultáneamente una cantidad
equivalente de cloruro o bromuro de hidrógeno. Hoy día se ha descubierto que se
obtienen resultados análogos con flúor, si se emplea diluido con un gas inerte
y en un equipo diseñado para extraer el calor producido. Al igual que en el
caso del metano, la yodación no tiene lugar.
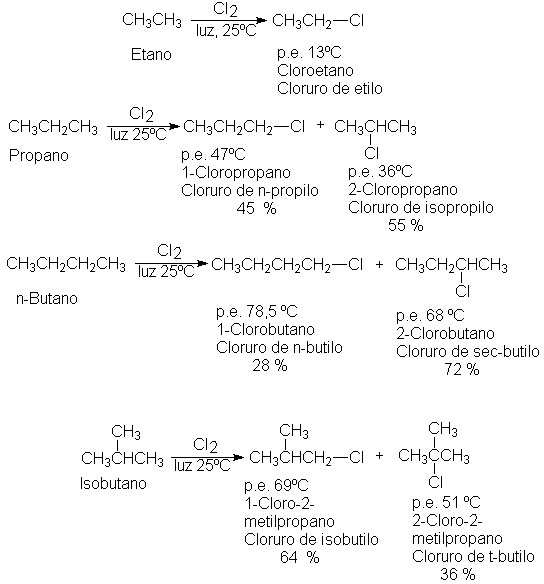
De un solo alcano puede generarse cualquiera de varios productos
isómeros, dependiendo del átomo de hidrógeno reemplazado. Así, el etano puede
dar un solo haloetano; el propano, el n-butano y el isobutano pueden generar
dos isómeros cada uno; el n-pentano, tres, y el isopentano, cuatro. Se ha
comprobado experimentalmente que al halogenar un alcano se forma una mezcla de
todos los productos isómeros posibles, lo que indica que todos los átomos de
hidrógeno son susceptibles al reemplazo. Para
la cloración, por ejemplo:
La bromación da los bromuros correspondientes, pero en proporciones diferentes:
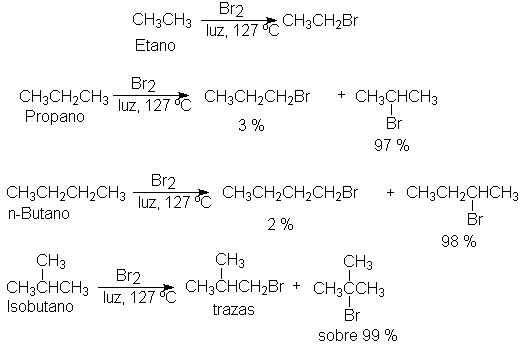
Aunque tanto la cloración como la bromación dan mezclas
de isómeros, los resultados expuestos
anteriormente indican que las cantidades
relativas de los distintos isómeros difieren mucho, dependiendo del halógeno usado. La cloración genera
mezclas en las que ningún isómero
predomina notoriamente; en cambio, en la bromación puede prevalecer un isómero
hasta el punto de constituir el único producto, con un 97-99% del total de la
mezcla. La bromación presenta alto
grado de selectividad en cuanto a los
átomos de hidrógeno que se reemplazan. (Como veremos en la Sec. 3.28, esta
característica de la bromación se debe a la reactividad relativamente baja de
los átomos de bromo y es un ejemplo de una relación general entre reactividad y selectividad.)
Con algunas excepciones, la halogenación de los alcanos no es apropiada para la preparación de
halogenuros de alquilo en el
laboratorio. En la cloración, cada producto se forma siempre con bajo rendimiento, y su separación de los
isómeros que lo acompañan es difícil, puesto que sus puntos de ebullición raras
veces son diferentes entre sí. La bromación de los alcanos casi no se utiliza.
En el capítulo 5 veremos que existen excelentes formas alternativas para obtener halogenuros de
alquilo en forma conveniente y a partir de precursores fácilmente disponibles.
Se empezó el estudio de las reacciones orgánicas con la
halogenación del metano y otros alcanos, no
por su utilidad como síntesis de laboratorio (la síntesis es solamente
un aspecto de la química orgánica), sino porque esta reacción ofrece un
acercamiento sencillo para la comprensión
de los principios subyacentes a todas las reacciones que estudiaremos.
Los alcanos son compuestos sencillos;
el mecanismo de la halogenación se conoce bien y está basado en
evidencias que podemos rápidamente. Podemos tratar rigurosa y cuantitativamente
las velocidades relativas y la orientación, puesto que se conocen con exactitud
los valores de Eact y DH. La naturaleza del
estado de transición está aclarada,
pero es incierto el papel que tiene el disolvente. Finalmente, el estudio de
los radicales libres es, por sí mismo, una parte importante de al química
orgánica.
A escala industrial, la cloración de alcanos es
importante. Para otros fines, como, por ejemplo, el empleo como disolvente, es
igualmente útil y mucho más barata una mezcla de isómeros que un compuesto
puro. Cuando la necesidad así lo indique, aun puede valer la pena la separación
de una mezcla de isómeros si cada isómero tiene mercado propio.
3.20 Mecanismo
del halogenación
La halogenación de los alcanos sigue el mismo mecanismo
que la del metano:
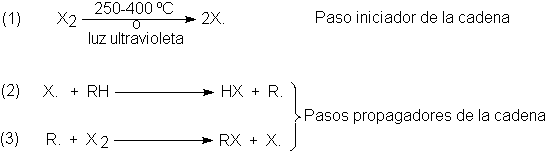
Un átomo de halógeno separa un hidrógeno del alcano (RH) para
formar un radical alquilo (R.), el cual, a su vez, quita un átomo de halógeno
de una molécula para dar el halogenuro de alquilo (RX).
El halogenuro de alquilo que se obtenga depende del
radical alquilo formado.
A su vez, esto depende del alcano y del hidrógeno que le
sea sustraído; por ejemplo, se obtiene el halogenuro de n-propilo de un radical
n-propilo, formado por separación de un hidrógeno primario del propano; el
halogenuro de isopropilo se obtiene del radical isopropilo, que se forma por separación
de un hidrógeno secundario.
La velocidad de formación del halogenuro de alquilo
depende de lo rápido que se forme el
radical alquilo. Al igual que en el caso del metano(Sec. 2.20), aquí también el
paso propagador (2) es más difícil que
el (3), por lo que controla la velocidad de la reacción total. La formación del
radical alquilo es difícil, pero una vez formado, se convierte con facilidad en
el halogenuro (véase Fig. 3.11).
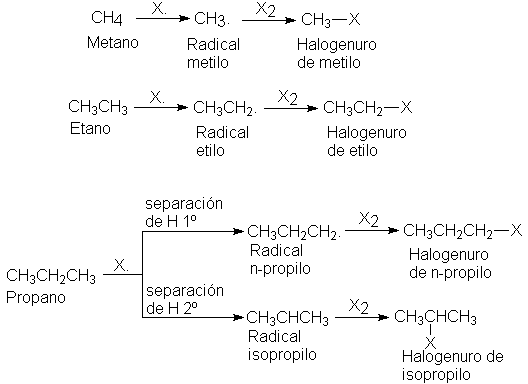
3.21 Orientación
de la halogenación
Con estos antecedentes, volvamos al problema de la orientación; es decir, examinemos los
factores que determinan en que lugar
de una molécula es más probable que se produzca la reacción. Este es un
problema que encontramos una y otra vez
al estudiar un compuesto que presente más de un lugar reactivo frente al ataque
de una sustancia. Se trata de un
problema importante, porque la orientación determina el producto que se
obtiene.
Tomemos como ejemplo la cloración del propano. Las
cantidades relativas de cloruro de
n-propilo e isopropilo que se obtienen dependen de las velocidades relativas de
formación de los radicales respectivos. Si se forman más rápidamente los
radicales isopropilo, también lo hará el cloruro de isopropilo, que la fracción
mayor del producto. Podemos apreciar
que los radicales n-propilo se forman por separación de hidrógenos
primarios, y los isopropilo, por separación de hidrógenos secundarios.
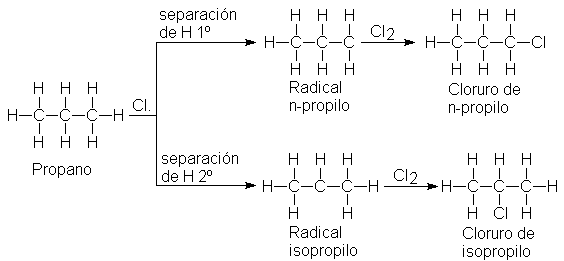
La
orientación queda determinada por la velocidad relativa de reacciones
competitivas. En este caso,
estamos comparando la velocidad de separación de hidrógenos primarios y
secundarios. ¿Cuáles son los factores
que determinan la velocidad de estas dos reacciones, y en cuál de ellos
puede diferir?
En primer lugar, tenemos la frecuencia de colisiones, que
debe ser igual para ambas reacciones,
puesto que las dos implican choques de las mismas partículas: una molécula de
propano y un átomo de cloro.
Luego está el factor de probabilidad. Si ha de separarse
un hidrógeno primario, la orientación de la molécula de propano, en el
instante de la colisión, debe ser tal
que el átomo de cloro golpee un hidrógeno primario; si ha de sustraerse uno
secundario, la orientación del propano debe ser tal que el cloro choque con uno
secundario. Puesto que hay seis hidrógenos primarios y solamente dos
secundarios, podemos estimar que el factor probabilidad favorezca la separación de hidrógenos
primarios en la proporción de 6:2 ó 3:1.
Considerando sólo la frecuencia de colisiones y nuestra
conjetura acerca de los factores de probabilidad, predecimos que la cloración
del propano da los cloruros de n-propilo e isopropilo en la proporción de 3:1.
Sin embargo, se indicó antes (Sec. 3.19) que ambos se forman en proporciones
similares, es decir, aproximadamente 1:1 ó 3:3. La cantidad de cloruro de
isopropilo es unas tres veces mayor que la predicha. Es evidente que las
colisiones con hidrógenos secundarios son tres veces más exitosas que con
hidrógenos primarios. Si nuestra posición
acerca del factor de probabilidad es correcta, significa que la Eact es menor para la separación de un hidrógeno
secundario que de uno primario.
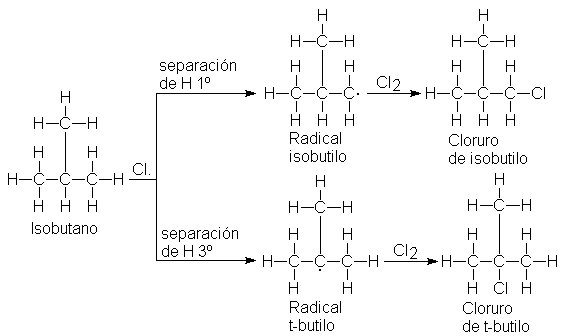
La cloración del isobutano presenta un problema similar. En este caso, la separación de uno
de los nueve hidrógenos primarios lleva a la formación del cloruro de
isobutilo, mientras que la separación
del único hidrógeno terciario conduce a la producción de cloruro de t-butilo.
En consecuencia estimaríamos que el
factor de probabilidad favorece la
formación del cloruro de isobutilo en la proporción de 9:1. Resultados
experimentales, expuestos en la sección
3.19, indican que dicha razón es aproximadamente 2:1 ó 9:45. Sin duda, las colisiones productivas son 4:5 veces más
numerosas con el hidrógeno terciario que con los primarios. A su vez, esto probablemente significa que la Eact
es menor para la separación de un hidrógeno terciario que de uno primario
y, de hecho, aún menor que para la separación de un hidrógeno secundario.
El estudio de la cloración de muchos alcanos ha demostrado
que éstos son resultados típicos. Considerando las diferencias en el factor de
probabilidad, se ha establecido que la velocidad de separación de átomos de
hidrógeno siempre sigue la secuencia 3º > 2º >1º; por
ejemplo, a temperatura ambiente la velocidad relativa por átomos de hidrógeno es 5.0:3.8:1.0. Empleando estos valores,
podemos predecir bastante bien la proporción de productos isómeros de la
cloración de un alcano dado; por ejemplo:
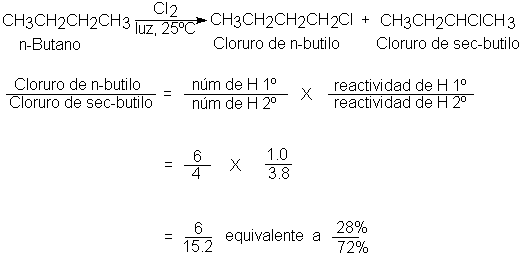
A pesar de estas diferencias en reactividad, raras veces
la cloración produce una preponderancia marcada de un isómero determinado. En
casi todo alcano, como en los ejemplos estudiados, son más numerosos los
hidrógenos menos reactivos, cuya menor reactividad se compensa con un factor de
probabilidad mayor, con el resultado de que se obtienen cantidades apreciables
de cada isómero.
Para la bromación se encuentra la misma secuencia de
reactividad, 3º > 2º > 1º, pero con
variaciones mucho mayores. A 127ºC, por ejemplo, la velocidad relativa por
átomos de hidrógeno es de 1600:82:1. En este caso, las diferencias de
reactividad son tan marcadas que prevalecen ampliamente sobre los factores de
probabilidad.
Parte 22
3.22 Reactividades relativas de los alcanos en la halogenación
La mejor manera de medir las reactividades relativas de
compuestos diferentes ante un mismo reactivo es por medio del método de competencia, ya que permite
una comparación cuantitativa exacta en condiciones idénticas de reacción. Se mezclan
cantidades equimolares de dos compuestos que van a ser comparados y se les hace
reaccionar con una cantidad limitada de
un reactivo determinado. Puesto que no hay suficiente reactivo para ambas sustancias, éstas compiten entre sí, y el
análisis de los productos de la reacción indica qué compuesto consumió
mayor proporción del agente, o sea, indica cuál es el más reactivo.
Si se hacen reaccionar cantidades equimolares de metano y
etano con una pequeña cantidad de cloro, por ejemplo, se obtiene unas 400 veces
más cloruro de etilo que de metilo, lo
que demuestra que el etano es 400 veces más reactivo que el metano.
Considerando el número de hidrógenos en las dos clases de moléculas, podemos
apreciar que cada hidrógeno del etano es unas 270 veces más reactivo que cada
hidrógeno del metano.
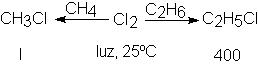
Los resultados
obtenidos en estudios similares con otros compuestos son compatibles con
la siguiente generalización sencilla: la
reactividad de un hidrógeno depende principalmente de su tipo, y no del alcano
al cual está unido; por ejemplo, cada hidrógeno primario del propano puede
separarse con la misma facilidad que cada hidrógeno primario del n-butano o
isobutano; cada hidrógeno secundario
del propano, tan fácilmente como los secundarios del n-butano o del n-pentano,
etc.
Los hidrógenos del metano corresponden a un tipo especial
y son aún menos reactivos que los primarios, como lo demuestra la competencia
con el etano.
3.23 Facilidad
de separación de átomos de hidrógeno. Energía de activación
Estamos en condiciones de resumir el efecto de la
estructura sobre la halogenación de los alcanos del siguiente modo. El paso que
controla la halogenación es la separación de hidrógeno por un átomo de
halógeno:
![]()
La facilidad relativa de separación de los distintos
tipos de átomos de hidrógeno es:
Facilidad de separación 3º > 2º > 1º > CH4
de átomos de hidrógeno
Esta secuencia es válida (a) para los diversos hidrógenos
en un mismo alcano, por lo que controla la orientación
de la reacción, y (b) para los hidrógenos de alcanos diferentes, por lo que
controla las reactividades relativas.
Ya antes habíamos sacado la conclusión de que estas
diferencias en la facilidad de separación -como la mayoría de las
diferencias en la velocidad de
reacciones íntimamente relacionadas (Sec. 2.19) - probablemente se deban a
diferencias en la Eact. Los valores
indicados en la tabla 3.5 se determinaron por estudios de halogenación a
varias temperaturas (Sec. 2.18). En concordia con nuestras conclusiones
tentativas, la velocidad creciente de reacción para la serie metilo, 1º, 2º, 3º
está en consonancia con una Eact decreciente. En la cloración, las
diferencias en Eact y en velocidad son pequeñas; en la bromación,
ambas son grandes.

Hemos visto (Sec. 2.18) que a mayor Eact de
una reacción, tanto mayor es el aumento de velocidad que produce un
incremento de la temperatura. Acabamos
de comprobar que las diferencias en la velocidad de separación de hidrógenos primarios, secundarios y terciarios
se deben a diferencias en la Eact. En consecuencia, predecimos que
un aumento de temperatura debe acelerar más la separación de hidrógenos
primarios (con Eact más alta) que la de hidrógenos terciarios (con Eact más baja),
por lo que los tres tipos de hidrógeno debieran exhibir una reactividad más
similar.
De hecho, este efecto nivelador ha sido observado: a
medida que la temperatura se eleva, la
velocidad relativa por átomo de
hidrógeno cambia de 5.0:3.8:1.0 hacia 1:1:1. A temperaturas muy altas,
virtualmente toda colisión tiene
energía suficiente incluso para separar hidrógenos primarios. Por lo general es
cierto que, a medida que se eleva la temperatura, un reactivo dado se
hace menos selectivo con respecto al
lugar de ataque; a la inversa, a medida que la temperatura disminuye, se
hace más selectivo.
¿Cómo podemos explicar el efecto de la estructura sobre
la facilidad de separación de átomos de hidrógeno? Puesto que esto es un asunto
de Eact, debemos buscar nuestra respuesta, como siempre, en el estado de
transición. Sin embargo, para hacerlo, debemos pasar nuestra atención de la
separación del átomo de hidrógeno al radical en formación.
3.24 Estabilidad
de radicales libres
En la tabla 1.2 (Sec. 1.14) hallamos las energías de
disociación homolítica de los enlaces que unen átomos de hidrógeno a varios grupos.
Estos son los valores DH de
las reacciones siguientes:
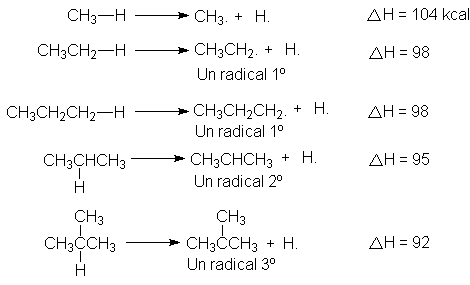
Por definición, la energía de disociación de enlace es la
cantidad de energía requerida para convertir un mol de alcano en radicales y
átomos de hidrógeno. Podemos apreciar que la energía requerida para la
formación de los distintos tipos de radicales decrece en el orden: CH3.
> 1º
> 2º > 3º.
![]()
Si se necesita menos energía para formar un radical que
otro, esto sólo puede significar que, en relación con el alcano que lo origina,
una radical contiene menos energía que el otro, es decir, es más estable (véase
Fig. 3.12).
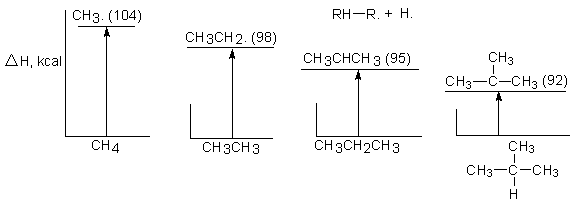
Fig.
3.12 Estabilidades relativas de radicales libres. (Gráficos alineados
entre
sí para simplificar la comparación.)
No intentamos comparar el contenido energético absoluto
de los radicales metilo o etilo, por ejemplo; simplemente observamos que la
diferencia en energía entre el metano y los radicales metilo e mayor que la
diferencia entre el etano y los radicales etilo. Cuando comparamos estabilidades relativas de radicales libre, debe
entenderse que nuestro patrón para cada radical es el alcano del cual se
deriva. Como veremos, es precisamente ésta la clase de estabilidad que nos
interesa.
Entonces, en relación con el alcano del cual se forma, el
orden de estabilidad de los radicales libres es:
Estabilidad 3º > 2º
> 1º
> CH3.
de radicales libres
3.25 Facilidad
de formación de radicales libres
Volvamos a la halogenación de los alcanos. Vimos que la
orientación y la reactividad (Sección 3.23) están
gobernadas por la facilidad relativa con que se separan las diferentes clases
de hidrógenos. Pero el hidrógeno que se separa y el radical que se forma
pertenecen, por definición, al mismo
tipo. La separación de un hidrógeno primario genera un radical primario; la de
uno secundario, un radical secundario, etc; por ejemplo:
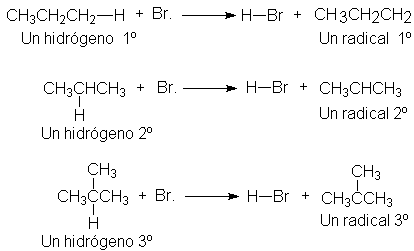
Si la separación de átomos de hidrógeno sigue el orden 3º > 2º >1º > CH4,
entonces la facilidad de formación de radicales libres debe seguir la misma
secuencia:
Facilidad de formación 3º > 2º
> 1º
> CH3.
de radicales libres
Al ordenar los radicales libres de acuerdo con la
facilidad de su formación, al mismo tiempo los hemos ordenado según su
estabilidad. Cuanto más estable sea un
radical libre, más fácilmente se forma.
Esta es una generalización muy útil. En muchas reacciones con formación de radicales libres, la estabilidad de
éstos parece dirigir la orientación y la reactividad. La adición de átomos
de bromo a los alquenos (Sec. 8.19), por ejemplo, es un tipo de reacción
diferente a la recién estudiada; sin embargo, en ella también se controla la
orientación y reactividad por la estabilidad de los radicales. (Aun en aquellos
casos en que son importantes otros factores, o incluso dominantes impedimento
estérico, efectos polares-, es conveniente emplear la estabilidad de los
radicales como punto de partida.)
3.26 Estado de
transición para la halogenación
¿Es
razonable que el radical más estable se forme con mayor facilidad?
Ya hemos visto que las diferencias en las reactividades
con átomos de halógeno se deben
principalmente a diferencias en la Eact. cuanto más estable
es el radical, menor es la Eact para su formación. A su vez, esto
significa que cuanto mayor se a la estabilidad de radical, más estable será el estado de transición que
conduce a su generación, midiéndose ambas
estabilidades, como debe ser, por comparación con el mismo patrón: los
reactivos. (Recuérdese: la Eact es
la diferencia del contenido energético
entre reactivos y estado de transición.)
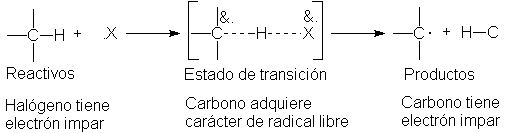
El examen del estado de transición demuestra que esto es
precisamente lo esperado. Como hemos visto antes (Sec. 2.23), el enlace
hidrógeno-halógeno está formado parcialmente, mientras que el enlace
carbono-hidrógeno está parcialmente escindido. El carácter de radical libre,
adquirido por el grupo alquilo, depende del grado de ruptura de dicho enlace:
los
factores que tienden a estabilizar al
radical libre, también tiende a
estabilizar al radical libre incipiente en el estado de transición.
Hemos visto que las estabilidades de radicales libres siguen la secuencia 3º > 2º> 1º> CH3. Por
ejemplo, cierto factor (deslocalización
del electrón impar, Sec. 10.11) hace que la diferencia de energía entre el isobutano y el radical
t-butilo sea menor que entre el propano y el radical isopropilo. Es razonable
que, en el estado de transición, este mismo factor haga que la diferencia
energética entre el isobutano y el
radical t-butilo incipiente sea menor
que entre el propano y el radical isopropilo incipiente (Fig. 3.13).
3.27 Orientación
y reactividad
En el estudio de al química orgánica, nos enfrentamos con
los problemas de orientación y reactividad del modo siguiente:
Ambos problemas implican la comparación de las
velocidades de reacciones íntimamente relacionadas: en el caso de la
orientación, se trata de reacciones en lugares diferentes del mismo compuesto;
en el caso de la reactividad, de reacciones con compuestos diferentes. Para
procesos tan íntimamente ligados, las variaciones en velocidad se deben
sobre todo a diferencias en la Eact que, por definición, es la
diferencia del contenido energético entre reactivos y estado de transición.
En consecuencia, examinaremos la estructura más probable
para el estado de transición, para ver qué características estructurales
afectan a su estabilidad sin afectar, al mismo
tiempo y en igual magnitud, a la estabilidad de los reactivos, es decir,
buscaremos factores que tienden a
aumentar o disminuir la diferencia energética entre reactivos y estado de transición. Una vez establecidas las
características estructurales que afectan
a la Eact, compararemos los estados de transición para las
reacciones cuya velocidad queremos
cotejar: cuanto más estable es el estado de transición, más veloz es la
reacción.
Tal como en el caso presente, en muchas reacciones con formación
de radicales libres, si no en todas, el estado de transición difiere de los
reactivos principalmente por el hecho de parecerse al producto. En
consecuencia, es razonable que el factor que más afecta a la Eact sea
el carácter de radical del estado de
transición. Por tanto, concluimos que, cuanto más estable sea el radical, más
estable será el estado de transición que conduce a él, y más rápidamente se
generará el radical.
3.28 Reactividad
y selectividad
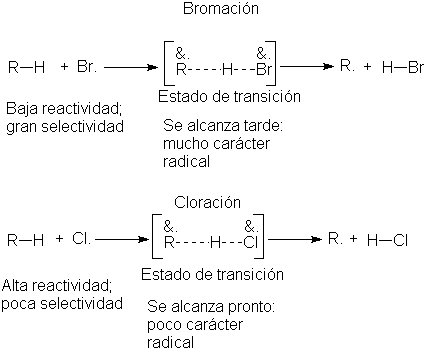
En su ataque a los alcanos, el átomo de bromo es mucho
más selectivo que el de cloro (con factores de velocidad relativa de 1600:82:1,
comparándolos con 5:0:3.8:1); también es mucho menos reactivo que el átomo de
cloro (frente al metano, por ejemplo, sólo 1/375 000 veces tan reactivo, como se vio en Sec. 2.19).
Este es un ejemplo de una relación general: en un conjunto de reacciones similares, una sustancia es
tanto más selectiva en su ataque,
cuanto menos reactiva sea.
Para explicar esta relación, debemos recordar lo
estudiado en la sección 2.24. Durante el proceso de ataque por el
comparativamente poco reactivo átomo de bromo, el estado de transición se
alcanza tarde en el proceso de la reacción, cuando el grupo alquilo ha
desarrollado un carácter radical considerable, mientras que, en el caso del
átomo de cloro muy reactivo, el estado de transición se alcanza pronto, cuando
el grupo alquilo aún no ha desarrollado mucho carácter radical.
Ahora bien, entendemos que <<selectividad>> se refiere a las diferencias
en velocidad de formación de los varios tipos de radicales libres; dijimos que
el más estable se forma más rápido, porque el factor que lo estabiliza la
deslocalización del electrón impar
(Sección 10.11)- también estabiliza al radical incipiente en el estado de
transición. Si esto es así; quiere decir
que cuanto más desarrollado se
encuentre el carácter radical en el
estado de transición, más efectiva será la deslocalización al estabilizar este último. Por ejemplo, el radical
isopropilo es 3 kcal más estable que el
n-propilo; si los radicales
estuvieran completamente formados en el estado de transición, la diferencia en
la Eact sería de 3 kcal. De hecho, en la bromación, esta diferencia
es 3 kcal: dentro de los límites de error experimental, igual a la
estabilización potencial máxima, lo que indica, como era de esperar, un
considerable carácter radical. Por el
contrario, en la cloración, la diferencia en la Eact es sólo 0.5
kcal, lo que indica muy poco carácter
radical.
Para reacciones de otro tipo, se presenta una situación
análoga. Cualquiera que sea el factor
responsable de las diferencias en estabilidad entre un conjunto de
estados de transición deslocalización de un electrón impar, o acomodación de
una carga positiva o negativa, o, quizá, un cambio en la aglomeración de los
átomos aquél aperará con más
efectividad cuando el estado de
transición se encuentre más desarrollado, es decir, cuando la sustancia sea
menos reactiva.
3.29 Ausencia
de transposición en los radicales libres. Trazadores isotópicos
Nuestra interpretación
de la orientación (Sec. 3.21) se basó en una
suposición que aún no hemos justificado: que las cantidades relativas de
halogenuros isómeros que encontramos en el producto reflejan la velocidad de
formación de los diversos radicales a partir del alcano correspondiente. Del isobutano, por ejemplo,
obtenemos dos veces más cloruro de isobutilo que de t-butilo, con lo que damos
por sentado que, por separación de hidrógeno, los radicales isobutilo se forman dos veces más
rápidamente que los t-butilo.
Sin embargo, ¿Cómo sabemos en este caso que cada radical
isobutilo que se forma da finalmente una molécula de cloruro de isobutilo?
Supongamos que algunos de estos
radicales cambiaran por transposición de átomos - a radicales
t-butilo, que reaccionarán luego con cloro para generar cloruro de t-butilo.
Esta suposición no es tan descabellada como podríamos pensar por nuestro
conocimiento actual. La duda a que da lugar es muy real; como veremos pronto,
otra partícula reactiva intermediaria, el carbocatión, es muy propensa a la
transposición: los iones menos estables
se transforman fácilmente en más estables (Sección 5.23).
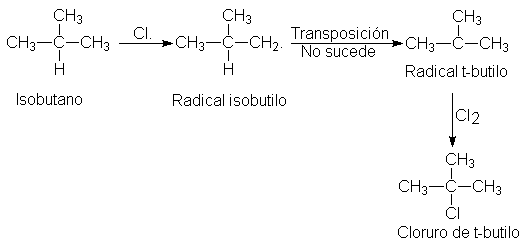
H. C. Brown (véase Sec. 17.11) y Glen Russell (ahora de
la universidad del Estado de Iowa) decidieron poner a prueba la posibilidad de
transposición de radicales libres, y eligieron la cloración del isobutano como
un buen ejemplo de prueba, debido a la gran diferencia de estabilidad entre los
radicales t-butilo e isobutilo. Si efectivamente es posible la transposición de radicales libres,
ciertamente debería suceder en este caso.
El problema se reduce a lo siguiente: ¿Conduce cada separación
de hidrógeno primario a cloruro de isobutilo, y cada separación de
hidrógeno terciario, a cloruro de t-butilo?
Podríamos suponer que sería imposible saberlo, puesto que
todos los átomos de hidrógeno son iguales; pero, ¿lo son efectivamente? En
realidad, existen tres isótopos del
hidrógeno: 1H o protio,
hidrógeno ordinario; 2H o D,
deuterio, hidrógeno pesado, y 3H o T, tritio. La distribución natural del protio y deuterio está en la relación 5000:1. (El
tritio, el isótopo inestable radiactivo, sólo se presenta en trazas, pero puede
hacerse por bombardeo del 6Li
con neutrones.) Métodos modernos de separación de isótopos permiten la
obtención de deuterio muy puro, a precios moderados, en forma de óxido de
deuterio, o agua pesada, D2O.
Brown y Russell prepararon el isobutano I marcado con
deuterio,
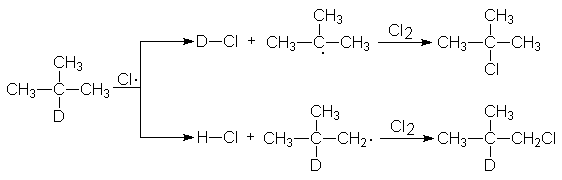
luego lo cloraron fotoquímicamente y analizaron los
productos, encontrando que la relación DCI:HCI (determinada con el
espectrómetro de masas) era igual a la del cloruro de t-butilo: cloruro de isobutilo
(dentro del margen de error experimental). Evidentemente, cada separación de
hidrógeno terciario (deuterio) dio
una molécula de cloruro de t-butilo y
cada separación de hidrógeno primario
(protio) dio una molécula de cloruro de isobutilo. No se produjo la transposición de radicales libres intermediarios.
Toda evidencia indica claramente que, aunque ocurren
transposiciones de radicales libres en ocasiones, éstas no son comunes y no
implican radicales alquilo simples.
El trabajo de Brown y Russell es sólo un ejemplo de cómo
podemos adquirir un conocimiento de una reacción química, empleando compuestos
marcados isotópicamente. Encontraremos muchos otros ejemplos en los que el
empleo de isótopos, ya como trazadores, como en este caso, ya para la detección
de efectos isotópicos (Sec. 7.17), nos da información acerca del mecanismo de
reacción, que no poderíamos obtener de ninguna
otra forma.
Aparte del deuterio y del tritio, en química orgánica se
utilizan a menudo otros isótopos que incluyen: 14C, disponible como 14CH3OH
y Ba14CO3; 18O, como H2 18O;
15N, como 15NH3,
15NO3- y 15NO2-;
36CI, como cloro o cloruro; 131I, como yoduro.
3.30 Combustión
La reacción de los alcanos con el oxígeno para formar
dióxido de carbono, agua y lo más importante calor, es la principal reacción que se desarrolla en una máquina de
combustión interna: su tremenda importancia práctica es evidente.
El mecanismo de esta reacción es muy complicado y aún no
se conoce bien. Sin embargo, parece indudable que se trata de una reacción en
cadena de radicales libres. Es un proceso
exotérmico y, sin embargo, para
su iniciación se requiere una temperatura muy
elevada, como la de una llama. Como en la cloración, se necesita mucha
energía para romper los enlaces que generan las partículas reactivas
iniciales; una vez vencida esta
barrera energética, los pasos
siguientes, propagadores de la cadena, proceden sin dificultad y con la
evolución de energía.
Un factor de comprensión más elevado ha hecho el motor de
gasolina más eficiente que los
antiguos, pero ha creado, al mismo tiempo, un problema: en ciertas condiciones,
la explosión suave de la mezcla combustible-aire es reemplazada en el cilindro
por un golpeteo que reduce
considerablemente la potencia del motor.
El problema del golpeteo ha sido resuelto en forma
satisfactoria de dos modos: (a) por
adecuada selección de los hidrocarburos que van a ser empleados como
combustible, y (b) por adición de tetraetilplomo.
Experimentos con compuestos puros han demostrado que los
hidrocarburos de estructuras diferentes difieren ampliamente en su tendencia al
golpeteo. La tendencia antigolpe relativa de un combustible se denomina
generalmente índice de octano. Se ha
establecido una escala arbitraria que le asigna al n-heptano, que golpea muy
fuerte, un índice de octano cero y al
2,2,4-trimetilpentano (<<iso-octano>>) un índice 100. Hoy se
dispone de combustibles con propiedades antigolpe superiores al iso-octano).
La fracción de gasolina
obtenida por destilación directa del petróleo (straighrun) se mejora por adición de compuestos de mayor índice de
octano; a veces, incluso, es reemplazada por estos combustibles mejores. Por lo
general, los alcanos ramificados, los alquenos y los hidrocarburos aromáticos
tienen excelentes cualidades antigolpes y se producen a partir de los
hidrocarburos del petróleo por medio del cracking
catalítico (Sec. 3.31) y la reformación
catalítica (Sec. 15.5). Los alcanos muy ramificados se sintetizan de
alquenos y alcanos por alquilación
(Sec. 8.17).
En 1922, T. C. Midgley, Jr., y T. A. Boyd (del
Laboratorio de Investigaciones de General
Motors) descubrieron que el índice de octano de un combustible puede
mejorarse considerablemente si se agrega una pequeña cantidad de tetraetilplomo, (C2H5)4Pb.
La gasolina así tratada se denomina
gasolina etílica o gasolina emplomada. Cerca de 50 años de
investigación han probado, finalmente, que el tetraetilplomo probablemente
funcione produciendo partículas
minúsculas de óxidos de plomo, sobre cuyas superficies se interrumpen
ciertas cadenas de reacciones.
Aparte del dióxido de carbono y el agua, el motor de
gasolina descarga a la atmósfera otras
sustancias, que son formadoras de contaminación o claramente venenosas: hidrocarburos no quemados, monóxido de
carbono, óxidos de nitrógeno y, de la gasolina emplomada, varios compuestos de plomo en Estados
Unidos, cientos de toneladas de plomo al día-. La inquietud pública creciente
en torno a estos contaminantes ha causado
una pequeña revolución en las
industrias del petróleo y del automóvil. Se están desarrollando convertidores para limpiar las emisiones
de los escapes, bien por oxidación catalítica de hidrocarburos y monóxido de
carbono, o por degradación de óxidos de nitrógeno en nitrógeno y oxígeno; pero la mayoría de estos catalizadores de
oxidación contienen platino, que es envenenado por el plomo. Se ha pensado en
eliminar el plomo de la gasolina, no en principio con el fin de reducir la
contaminación plúmbica, sino para permitir que funcionen los convertidores.
Esto ha resucitado, a su vez, el problema del golpeteo, que se está enfrentando
de dos formas: (a) reduciendo el factor de comprensión de los motores de los
nuevos automóviles, y (b) aumentando el
índice de octano de la gasolina por cambios en su composición de hidrocarburos,
con adición de aromáticos y mayor uso de la isomerización (Sec.
3.13).
3.31 Pirólisis:
cracking
La descomposición de una sustancia por la sola acción del calor, se denomina pirólisis (del griego: pyr,
<<fuego>>, y lysis, <<pérdida>>), lo que los químicos entienden por <<descomposición por el
calor>>;
compárese con hidrólisis, <<descomposición por el
agua>>
La pirólisis de alcanos, en particular en lo que
concierne al petróleo, se conoce como cracking. En el cracking térmico, los alcanos simplemente se hacen pasar por una
cámara calentada a temperatura elevada: los alcanos pesados se convierten en
alquenos, alcanos livianos y algo de hidrógeno. Este proceso produce
predominante etileno (C2H4), junto con otras moléculas
pequeñas. En una modificación, llamada cracking
al vapor, se mezclan los
hidrocarburos con vapor, se calientan a 700-900ºC por fracción de segundo y se
enfrían rápidamente. Este proceso está
adquiriendo importancia creciente en la producción de hidrocarburos para
síntesis, incluyendo etileno, propileno, butadieno, isopreno y ciclopentadieno.
Otra fuente de hidrocarburos menores es el hidrocracking,
que se desarrolla en presencia de hidrógeno a presión alta y a temperaturas
mucho más bajas (250-450ºC).
Los alquenos de bajo peso molecular obtenidos por estos
procedimientos pirolíticos pueden separarse y purificarse, y son las materias primas
más importantes para la síntesis a gran escala de compuestos alifáticos.
Sin embargo, la mayor parte de la pirólisis va dirigida a
la producción de combustibles y no a la producción de materias primas, siendo
para aquellos el proceso más importante el
cracking catalítico. Fracciones más pesadas del petróleo (típicamente,
gasóleo) se ponen en contacto con un catalizador de sílice-alúmina finamente
dividido a 450-550ºC, y bajo una ligera
presión. Este proceso no sólo aumenta la producción de gasolina, rompiendo moléculas grandes en otras más pequeñas,
sino que también mejora su calidad: el método
involucra carbocationes (Sec. 5.17) y genera alcanos y alquenos con las
estructuras altamente ramificadas que se desean para la gasolina.
Por medio del proceso de alquilación (Sec. 8.17), algunos de los alcanos menores y los alquenos se convierten en
combustibles sintéticos de alto octanaje.
Finalmente, se convierten cantidades enormes de
hidrocarburos alifáticos del petróleo en hidrocarburos aromáticos, por medio del proceso de reformación catalítica (Sec. 15.5), que
no sólo se emplean como combustibles de calidad superior, sino, también, como
materias primas para síntesis de la mayoría de los compuestos aromáticos (Cap.
13).
3.32 Determinación
de la estructura
En química orgánica, una de las tareas más comunes e
importantes es determinar la fórmula estructural de un compuesto recién
sintetizado o aislado de una fuente natural.
La sustancia
pertenecerá a uno de dos grupos, aunque en un comienzo probablemente no
sabremos a cuál de ellos: puede tratarse (a) de un compuesto descrito
previamente, el cual debemos identificar, o (b) de un material nuevo, cuya
estructura debemos determinar.
Si el compuesto ya ha sido localizado por algún químico,
que ya ha determinado su estructura, entonces se encontrará una descripción de
sus propiedades en la bibliografía química, junto con las pruebas que sirvieron
de base para asignarle la estructura. En tal caso, sólo se necesita demostrar que
nuestra sustancia es idéntica a la ya descrita.
Por el contrario, si nuestro compuesto es nuevo y no ha
sido descrito antes, deberemos desarrollar una prueba estructural mucho más
elaborada.
Veamos por ahora
de una forma general y luego en más detalle cómo atacaríamos este
problema.
Tenemos un matraz lleno de un gas, unos pocos mililitros
de un líquido o un montoncito minúsculo de cristales. Debemos hallar respuesta
a la pregunta: ¿qué es?
En primer lugar, purificamos la sustancia y determinamos
sus propiedades físicas: puntos de
fusión y ebullición, índice de refracción, solubilidad en varios disolventes.
En un laboratorio moderno, mediríamos
varios espectros del compuesto (Cap. 16), en particular, el infrarrojo y el de RMN; de hecho, debido al
gran caudal de información que puede obtenerse de este modo, el examen
espectroscópico suele ser el primer trabajo después de la purificación. El espectro de masas nos daría un peso
molecular muy preciso. Cada vez es más frecuente determinar la estructura del
modo más directo posible: mediante el análisis con rayos X, que puede indicar
la distribución precisa de los átomos en una molécula.
Realizaríamos un análisis cualitativo elemental para ver
qué elementos están presentes (Sec.
2.26). Podríamos seguir con un análisis cuantitativo, con el que, combinado con
el peso molecular, obtendríamos una fórmula molecular (Sec. 2.27); ciertamente
haríamos esto, si sospecháramos la presencia de una sustancia nueva.
A continuación, estudiamos sistemáticamente el comportamiento
del compuesto frente a ciertos reactivos, lo que, combinado con el análisis
elemental, las propiedades de solubilidad y los espectros, nos permite caracterizar la sustancia, es decir,
decidir a qué familia pertenece.
Por ejemplo, podríamos encontrar que se trata de un
alcano, un alqueno, un aldehído o un éster.
La pregunta siguiente es: ¿de qué alcano se trata? O ¿de qué alqueno, aldehído o éster?
Para encontrar la respuesta, nos dirigimos primero a la
literatura química y buscamos datos de compuestos que pertenezcan a la misma
familia de nuestro desconocido.
Si encontramos descrito uno que tenga las mismas
propiedades físicas que la sustancia desconocida, las probabilidades de que
ambas sean idénticas, son buenas. Para
confirmarlo, generalmente convertimos
la desconocida por medio de una reacción química en un nuevo compuesto, llamado
derivado, y demostramos que este
derivado es idéntico al producto obtenido en la misma forma del compuesto
previamente descrito.
Por el contrario, si no encontramos la descripción de una
sustancia cuyas propiedades físicas sean idénticas a las de nuestra sustancia,
nos enfrentamos con una difícil tarea: tenemos un compuesto nuevo y debemos
comprobar su estructura. Podemos realizar una
degradación, o sea, desmenuzar la molécula, identificar los fragmentos y
deducir cuál debe haber sido la
estructura. Para confirmar toda prueba estructural, intentamos sintetizar
la sustancia desconocida por un método
que no deje dudas acerca de su estructura.
En el capítulo 16, y una vez que nos hayamos
familiarizado con más aspectos de las estructuras orgánicas, veremos cómo
encaja la espectroscopia en el procedimiento general descrito antes.
3.33 Análisis
de los alcanos
Un compuesto desconocido se caracteriza como alcano por
pruebas negativas.
El análisis elemental cualitativo de un alcano da
resultados negativos para todos los
elementos, excepto el carbono y el hidrógeno. Una combustión
cuantitativa, si se efectúa, demuestra la ausencia de oxígeno; junto con una
determinación del peso molecular, la combustión da la fórmula molecular, CnH2n+2,
que corresponde a un alcano.
Un alcano no sólo es insoluble en agua, sino también en
ácidos y bases diluidos, y en ácido
sulfúrico concentrado. (Como veremos, la mayoría de los compuestos orgánicos
se disuelven en uno o más de estos
disolventes.)
Un alcano es inerte frente a la mayoría de los reactivos
químicos. Su espectro infrarrojo carece de las bandas de absorción
características de los grupos atómicos presentes en otras familias de
sustancias orgánicas (como OH, C=O, C=C, etcétera).
Una vez que el compuesto
ha sido identificado como alcano, aún falta la segunda parte del problema: descubrir qué alcano es.
Partiendo de sus propiedades físicas puntos de ebullición
y fusión, densidad, índice de
refracción, espectros infrarrojo y de masas, que son los más confiables
puede ser identificado como un alcano previamente descrito y de estructura
conocida.
Si resulta ser un alcano nuevo, la comprobación
estructural puede ser tarea difícil. La
combustión y determinación del peso molecular dan su fórmula molecular. Sus
espectros infrarrojos y de RMN dan pistas sobre la disposición de sus átomos.
(Para compuestos como los alcanos, puede resultar necesario apoyarse
fuertemente en la difracción de rayos X y la espectrometría de masas.)
La comprobación final está en la síntesis del compuesto
desconocido por un método que sólo puede conducir a la estructura específica
asignada.