Metano
(Ir a página principal http://organica1.org/ )
2.6 Oxidación. Calor de combustión
2.7 Cloración: una reacción de sustitución
2.9 Reacción con otros halógenos: halogenación
2.12 Mecansimo de la cloración.
2.17 Avance de la reacción: cambios de energía
2.19 Velocidad relativa de reacción
2.20 Reactividades relativas de los halógenos con el
metano
2.21 Mecanismo alternativo para la halogenación
2.22 Estructura del radical metilo. Hibridación sp2
2.24 Reactividad y desarrollo del estado de transición
2.25 Fórmula molecular: su importancia fundamental
2.26 Análisis elemental cualitativo
2.27 Análisis elemental cuantitativo: carbono, hidrógeno
y halógeno
2.29 Peso molecular. Fórmula molecular
2.1 Hidrocarburos
Ciertos compuestos orgánicos sólo contienen dos
elementos, hidrógeno y carbono, por lo que se conocen como hidrocarburos. Partiendo de su estructura, se dividen en dos clases
principales: alifáticos y aromáticos. Los primeros se
subdividen en familias: alcanos, alquenos, alquinos y sus análogos cíclicos
(cicloalcanos, etc.)
Hidrocarburos
__________________½_______________
½
½
Alifáticos
Aromáticos
____
___________½_____________________
½ ½ ½ ½
Alcanos Alquenos Alquinos Alifáticos
cíclicos
El miembro más simple de la familia de los
alcanos, y de hecho uno de los compuestos orgánicos más simples, es el metano, CH4. Sólo
estudiaremos este compuesto con algún detalle, ya que todo lo que aprendemos
acerca de él puede aplicarse, con ligeras modificaciones, a cualquier alcano.
2.2 Estructura del metano
Como ya estudiamos
anteriormente (sec. 1.11), cada uno de los cuatro átomos de hidrógeno
está unido al de carbono por un enlace covalente, es decir, compartiendo un par de electrones.
Cuando el carbono está unido a otros cuatro átomos, sus orbitales enlazantes
(orbitales sp3, formados por mezcla de un orbital s y tres p) se
dirigen hacia los vértices de un tetraedro (Fig. 2.1a). Esta disposición
tetraédrica es lo que permite a los orbitales estar separados al máximo. Para
que cada uno de estos orbitales solape
al orbital esférico s de un átomo de hidrógeno con efectividad máxima, formando
así un enlace más fuerte, cada núcleo de hidrógeno debe ubicarse en un vértice
de este tetraedro (Fig. 2.1b).
La estructura tetraédrica del metano ha sido
verificada por difracción de electrones (Figura 2.1c), lo que muestra fuera de toda duda la disposición
de los átomos en moléculas tan simples. Más adelante analizaremos algunas
de las pruebas que permitieron la mecánica cuántica o la difracción de
electrones.
Normalmente, representaremos al metano con una
raya por cada par de electrones compartido por el carbono y el hidrógeno (I).
Para concentrar nuestra atención sobre electrones individuales, indicaremos
algunas veces un par por dos puntos (II); finalmente, para representar la forma
verdadera de la molécula, emplearemos una fórmula tridimensional simple
como III o IV.
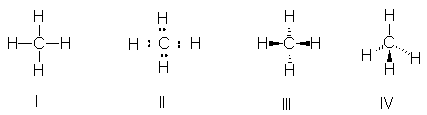
Fig 2.1
En las fórmulas
tridimensionales de este tipo, una cuña negra representa un enlace que sale del
plano del papel hacia nosotros; una cuña de puntos, un enlace en que se aleja
de nosotros por detrás del plano del papel, y una línea normal, un enlace en el
plano del papel. Por consiguiente, las fórmulas II y IV representan al metano,
como en las figuras 2.2a y 2.2b, respectivamente.
2.3 Propiedades físicas
Como vimos en el capítulo anterior (Sec. 1.18), la
unidad de un compuesto no iónico, sea sólido, líquido o gaseoso, es la molécula.
Como la molécula de metano es muy simétrica, las polaridades de los enlaces
carbono-hidrógeno individuales se anulan, de lo que resulta que la molécula en
sí no es polar.
La atracción entre tales moléculas no polares
queda limitada a las fuerzas de Van der Wasls; para moléculas tan pequeñas,
estas fuerzas atractivas deben ser muy débiles, comparadas con las intensísimas entre iones sodio y
cloruro, por ejemplo. No debe ser sorprendente, por tanto, que esas fuerzas atractivas
sean vencidas con facilidad por
la energía térmica, de modo que la fusión
y ebullición se producen a
temperaturas muy bajas: p.f. -183ºC, p.e. -161.5ºC. (Compárense estos
valores con los correspondientes para el cloruro de sodio: p.f. 801ºC, p.e.
1413ºC.) En consecuencia, el metano es un gas a temperatura ordinarias.
El metano es incoloro y, en estado líquido, menos
denso que el agua (densidad relativa 0.4); de acuerdo con la regla de que <<una
sustancia disuelve a otra similar>>, es apenas soluble en agua, pero muy soluble en líquidos orgánicos, como
gasolina, éter y alcohol. Con respecto a sus propiedades físicas, el metano
fija la pauta para los demás miembros de la familia de los alcanos.
2.4 Fuente
El metano es un producto final de la putrefacción
anaeróbica (sin aire) de las plantas,
es decir, de la descomposición de ciertas moléculas muy complejas. Como tal, es
el principal constituyente (hasta un 97%) del gas natural. Es el peligroso
grisú de las minas de carbón y pueden
verse aflorar burbujeando en las ciénegas
como gas de los pantanos.
Si se quiere metano muy puro, puede separarse por
destilación fraccionada de los otros constituyentes del gas natural (también
alcanos en su mayoría); la mayor parte se consume como combustible sin
purificar.
De acuerdo con una teoría, los orígenes de la vida
se remontan a una tierra primitiva, rodeada por una atmósfera de metano, agua,
amoniaco e hidrógeno. La energía -radiación del sol, descargas de relámpagos
rompió estas moléculas simples en fragmentos reactivos (radicales libres, Sec.
2.12), que se combinaron para formar
moléculas más grandes y finalmente dieron origen a los enormemente complejos
compuestos orgánicos que conforman los organismos vivos. (El descubrimiento de
moléculas orgánicas en el espacio ha llevado incluso a la especulación de
que <<en las
nubes interestelares pudieron existir
las semillas orgánicas para la vida>>.)
En 1953, en la Universidad de Chicago, el ganador
del Premio Nobel, Harold C. Urey, y su
colaborador, el estudiante Stanley Miller, encontraron pruebas de que esto pudo
haber sucedido. Demostraron que una descarga
eléctrica convierte una mezcla de
metano, agua, amoniaco e hidrógeno en un gran número de compuestos orgánicos,
incluso aminoácidos, que son los principios a partir de los cuales se forman
las proteínas, la sustancia de la vida (Cap. 40). (Es quizá lo más
apropiado comenzar nuestro estudio de la química orgánica con el metano y su conversión en radicales
libres.)
El metano generado en la descomposición final de
un organismo que alguna vez estuvo vivo, bien puede ser la sustancia a partir
del cual en última instancia se haya derivado ese organismo. <<La tierra a la tierra, las cenizas a las cenizas, el polvo al polvo.>>
2.5 Reacciones
Por sus
propiedades químicas y físicas, el
metano fija la norma para la familia de los alcanos (Sec. 3.18). Típicamente,
sólo reacciona con sustancias muy reactivas, o en condiciones muy vigorosas, lo
que redunda, como veremos, en lo mismo. De momento, sólo consideremos su oxidación:
por oxígeno, halógenos e, incluso, agua.
REACCIONES DEL METANO
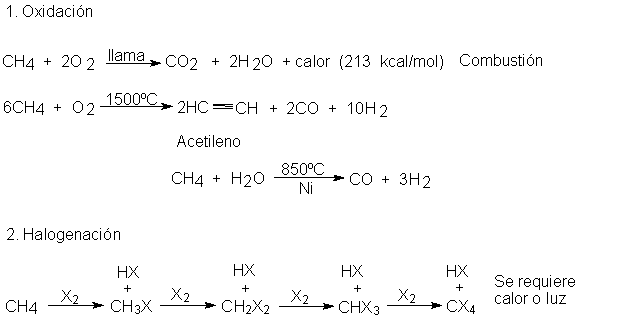
Reactividad de X2
F2 > CI2 > Br2 ( > I2)
2.6
Oxidación. Calor de combustión
La combustión a dióxido de carbono y agua es
característica de los compuestos orgánicos; en condiciones especiales, se emplea
para determinar su contenido en carbono
e hidrógeno (Sec. 2.27).
La combustión del metano es la reacción principal que tiene lugar al quemar gas
natural. Es casi innecesario hacer hincapié en su importancia en áreas donde se
dispone de este gas; el producto importante no es el dióxido de carbono ni el
agua, sino el calor.
La combustión de hidrocarburos sólo se efectúa a
temperaturas elevadas, como las que proporcionan una llama o una chispa. Sin
embargo, una vez iniciada, la reacción desprende calor, que a menudo es suficiente para
mantener la alta temperatura y permitir que la combustión continúe. La cantidad
de calor que se genera al quemar un mol de un hidrocarburo a dióxido de
carbono y agua se llama calor de
combustión: para el metano es 213 kcal.
La oxidación parcial controlada y su reacción
catalítica con agua a temperatura
elevada, convierte al metano en una fuente cada vez más importante de
otros productos que no sean calor: de
hidrógeno, empleado en la fabricación del amoniaco; de mezclas de monóxido de
carbono e hidrógeno, usadas en la síntesis del metanol y otros alcoholes; y del acetileno (Sec. 11.5), que, a su vez, es el punto de partida para la
producción a gran escada de muchos compuestos orgánicos.
La oxidación por halógenos es de interés especial
para nosotros en parte, porque sabemos
más de ésta que de las otras reacciones del metano y, en un sentido u otro, es
el tema de estudio del resto del capítulo.
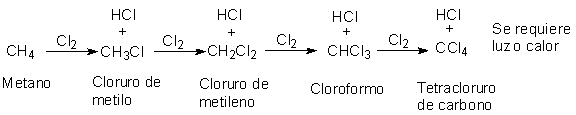
2.7 Cloración: una reacción de sustitución
Una mezcla gaseosa de metano y cloro reacciona
vigorosamente por influencias de la luz
ultravioleta o a una temperatura de 250-400ºC para producir cloruro de hidrógeno y un compuesto de
fórmula CH3CI. Se dice que el metano ha sufrido una cloración, llamándose el producto, CH3CI,
clorometano o cloruro de metilo (CH3=metilo).
La cloración es un ejemplo típico de una amplia
clase de reacciones orgánicas conocida como sustitución. Se ha sustituido un átomo de hidrógeno del metano por
uno de cloro, y el átomo de hidrógeno así reemplazado termina combinado con un
segundo átomo de cloro.
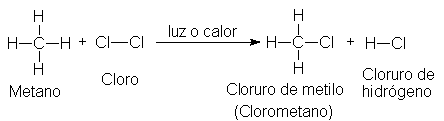
A su vez, el cloruro de metilo puede sufrir una
sustitución posterior, formando más cloruro de hidrógeno y el compuesto CH2CI,
diclorometano o cloruro de metileno
(CH2 = metileno).
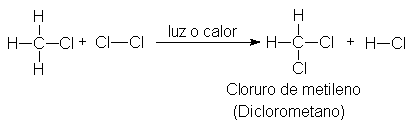
De modo análogo, la cloración puede continuar para
dar CHCI3, triclorometano o cloroformo, y CCI4, tetraclorometano o tetracloruro de carbono.
Este último fue en tiempos muy utilizados, pero ha sido susitutido casi
totalmente por otros materiales.
2.8 Control de la Cloración
La cloración del metano puede dar cualquiera de
cuatro productos orgánicos, dependiendo de hasta dónde se lleve la reacción. ¿Podemos controlarla de modo que
el principal producto orgánico sea el
cloruro de metilo? Es decir, ?Podemos limitarla a la primera etapa, la
monocloración?
Podríamos suponer ingenuamente, como se observará,
que esto se logra con sólo proporcionar un mol de cloro por cada mol de
metano. Veamos, sin embargo, lo que
sucede si procedemos así. Al comenzar la reacción, sólo hay metano para que
reaccione con el cloro, por lo que sólo
se realiza la primera etapa de la cloración; no obstante, este proceso
general cloruro de metilo, de modo que
al proseguir la reacción desaparece el metano y es reemplazado por cloruro de
metilo.
A medida que crece la proporción de cloruro de
metilo, éste compite con el metano por el cloruro. Cuando la concentración de
cloruro de metilo supera a la de metano, se hace más probable que el cloro
ataque al cloruro de metilo que al metano,
por lo que pasa a ser más importante la segunda etapa de la cloración que la primera. Se forma gran cantidad de
cloruro de metileno, que se convierte de forma análoga en cloroformo y
éste, a su vez, en tetracloruro de carbono. Finalmente, cuando procedemos a
aislar el producto de la reacción,
encontramos que es una mezcla de
los cuatro metanos clorados junto con algo de metano sin reaccionar.
Sin
embargo, puede limitarse la reacción casi exclusivamente a la
monocloración si empleamos un gran exceso de metano. En este caso, aun final
del areacción, el metano sin reaccionar supera en mucho al cloruro de metilo.
El cloro tiene mayor probabilidad de atacar al primero que al segundo, por lo
que la reacción principal es la primera etapa de la cloración.
Debido a la gran diferencia en sus puntos de
ebullición, es fácil separar el exceso de metano (p.e. 161.5ºC) del cloruro de
metilo (p.e. -24ºC), de modo que se puede mezclar el metano con más cloro y
someterlo nuevamente al proceso. Aunque la conversión del metano en cloruro de
metilo es baja en cada ciclo, La producción de cloruro de metilo a aprtir del
cloro consumido es bastante elevada.
El empleo de un gran exceso de un compuesto es un
recurso habitual para el químico
orgánico cuando quiere limitar la reacción a sólo uno de los varios
sitios reactivos en la molécula de esa sustancia.
2.9 Reacción con otros halógenos: halogenación
El metano reacciona con bromo, una vez más a
temperaturas elevadas o bajo la influencia de la luz ultravioleta, para dar los
correspondientes bromometanos: bromuro de metilo, bromuro de metileno,
bromoformo y tetrabromuro de carbono.

La bromación se realiza con menor facilidad que la
cloración.
El metano no reacciona con el yodo. Con el flúor,
la reacción es tan vigorosa que, aun en la oscuridad y a temperatura ambiente,
el proceso debe ser controlado cuidadosamente: los reactivos, diluídos con un
gas inerte, se mezclan a presión reducida.
En consecuencia, podemos ordenar los halógenos de
acuerdo con su reactividad:
Reactividad de los halógenos
F2 > CI2 > Br2 ( > I2)
Este mismo orden de reactividad vale para la
reacción de los halógenos con otros alcanos y, de hecho, con la mayoría de los compuestos
orgánicos. La distribución de las reactividades es tan amplia que solamente la
cloración y la bromación tiene lugar a velocidades que son de utilidad general.
2.10 Reactividad relativa
A lo largo de nuestro estudio de la química
orgánica se tratarán constantemente las relativas
reactividades. Compararemos las reactividades de diversas sustancias con un
mismo compuesto orgánico; las reactividades de distintos compuestos orgánicos
con un mismo reactivo, e incluso las reactividades de sitios diferentes de una
misma molécula orgánica con el mismo reactivo.
Cuando comparamos
reactividades debe entenderse que estamos comparando velocidades de
reacción. Cuando decimos que el cloro es más reactivo con el metano que el bromo, entendemos que, en las mismas
condiciones (igual concentración, igual temperatura, etc.), el cloro reacciona más rápidamente con el metano que el
bromo. Desde otro punto de vista, queremos decir que la reacción con bromo debe
realizarse en condiciones más vigorosas (concentración mayor o temperatura más
elevada) para que proceda a la misma velocidad que la cloración. Cuando decimos
que el metano y el yodo no reaccionan, entendemos que la reacción es demasiado
lenta como para ser significativa.
No sólo queremos saber cuáles son estas
reactividades relativas, sino también, dentro de lo posible, cómo
justificarlas. Para ver qué factores dan mayor velocidad a una reacción que a
otra, estudiaremos con más detalle las diferentes reactividades de los
halógenos con el metano. Sin embargo, antes de poder hacer esto, debemos
comprender mejor la propia reacción.
2.11 Mecanismos de reacción
No sólo es importante saber qué sucede en una
reacción química, sino también cómo
sucede, es decir, conocer no sólo los hechos,
sino también la teoría.
Por ejemplo, sabemos que, bajo la influencia del
calor o la luz, el metano y el cloro forman cloruro de metilo y cloruro de
hidrógeno. ¿Cómo exactamente, se convierte una molécula y de metano en otra de
cloruro de metilo? ¿Implica esta reacción más de un paso? Y en ese caso,
¿Cuáles son los pasos? ¿Cuál es la función del calor o de la luz?
La respuesta a tales preguntas, es decir, la descripción detallada, paso a paso, de
una reacción química se denomina mecanismo. Es solamente una hipótesis
propuesta para explicar los hechos. A mediad que se descubren nuevos hechos, el
mecanismo también debe explicarlos o debe ser modificado para incluirlos;
incluso puede ser necesario descartar un mecanismo para reemplazarlo por otro.
Sería difícil sostener que alguna vez haya sido demostrado un mecanismo. Sin
embargo, si explica satisfactoriamente
una amplia gama de hechos, si nos permite hacer predicciones que luego se cumplen, si es consistente con
mecanismos para otras reacciones relacionadas,
entonces se dice que tal mecanismo está bien fundado y pasa a formar parte de la teoría de la química orgánica.
¿Por qué nos interesan los mecanismos de las
reacciones? Como parte importante de la teoría de la química orgánica, contribuyen a erigir el marco
dentro del cual colocamos los hechos que observamos. La comprensión de los
mecanismos nos facilitará el descubrimiento de un patrón en la compleja y
desconcertante maraña de las reacciones orgánicas. Descubriremos que muchas reacciones, aparentemente sin
relación, tienen lugar por el mismo mecanismo o por mecanismos análogos, de
modo que la mayor parte de lo aprendido sobre una reacción puede aplicarse
directamente a muchas otras.
Sabiendo cómo tiene lugar una reacción, podemos
modificar las condiciones experimentales no por tanteo, sino lógicamente para que mejore el rendimiento del producto
que nos interesa o para cambiar por concepto el curso de la reacción y obtener
un resultado diferente. A medida que aumenta nuestro conocimiento de las
reacciones, también aumenta nuestra capacidad para controlarlas.
2.12 Mecansimo de la cloración.
Radicales libres
Será útil
examinar en detalle el mecanismo de la cloración del metano, que también es
válido para la bromación y la cloración de otros alcanos; también vale para muchos
compuestos que, aun no siendo alcanos, contienen partes similares a ellos en
sus moléculas. Mecanismos íntimamente relacionados se hallan involucrados en la
oxidación (combustión) y otras reacciones
de los alcanos. Es más, este mecanismo ilustra ciertos principios
generales que pueden traspasarse a una amplia gama de reacciones químicas. Por
último, al considerar los hechos que
apoyan este mecanismo, podemos aprender algo acerca de cómo el químico descubre
lo que sucede durante una reacción química.
Entre los hechos que deben ser considerados, se
destacan los siguientes:
(a) En la oscuridad, el metano y el cloro no
reaccionan a temperatura ambiente.
(b) Sin embargo, a temperaturas superiores a los
250ºC, la reacción procede con facilidad en la oscuridad, o
(c) a temperatura ambiente por influencia de la
luz ultravioleta.
(d) La longitud de onda de la luz que induce la
cloración es la que se sabe que causa, independientemente, la disociación de
moléculas de cloro.
(e) Cuando se induce la reacción con luz, se
obtiene muchas moléculas (varios miles) de cloruro de metilo por cada fotón
absorbido por el sistema.
(f) La presencia de una pequeña cantidad de
oxígeno frena la reacción por un periodo de tiempo, al cabo del cual ella
procede normalmente; la longitud de este
periodo depende de la cantidad de oxígeno del sistema.
(Daremos más evidencias sobre el mecanismo en las
Secs. 2.21 y 4.28.)
El mecanismo que mejor satisface estas
observaciones, por lo que goza de aceptación general, se muestra en las siguientes
ecuaciones:

El primer paso lo constituye la disociación de una
molécula de cloro en dos átomos. Al igual que la ruptura de cualquier enlace,
esto necesita energía, la energía de
disociación de enlace; de la tabla 1.2 (Sec. 1.14) se desprende que en este
caso el valor es de 58 kcal/mol. La energía se aporta en forma de luz o calor.
![]()
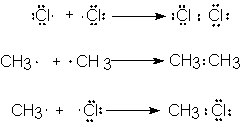
La molécula de cloro sufre homólisis (Sec. 1.14); es decir, la ruptura del enlace cloro-cloro
se produce simétricamente, de modo que cada átomo retiene un electrón del par
que constituía el enlace covalente. Este electrón
impar no está apareado como el resto de los electrones del átomo de cloro,
o sea, no tiene compañero con espin opuesto (Sec. 1.6). Un átomo o grupo de átomos que posee un electrón impar ( no apareado)
se denomina radical libre.
Al escribir el símbolo de un radical libre,
generalmente se incluye un punto para representar al electrón impar, igual que
un signo más o menos forma parte del símbolo de un ión.
Una vez formado, ¿qué es lo que probablemente le
sucederá al átomo de cloro? Como la mayoría de los radicales libres, éste es extremadamente reactivo,
debido a que tiende a adquirir un electrón adicional para completar así su
octeto; desde otro punto de vista, a cada átomo de cloro se le aportó energía
durante la ruptura de la molécula, por lo que esta partícula muy energética
tiene una fuerte tendencia a perder energía mediante la formación de un nuevo
enlace químico.
Para formar un
nuevo enlace, o sea, para reaccionar, el átomo de cloro debe chocar con algún
otro átomo o molécula. ¿Con qué tiene mayor probabilidad de chocar? Obviamente,
con las partículas presentes en mayor concentración: las moléculas de cloro y
de metano. La colisión con otro átomo de cloro es muy improbable, simplemente
porque en cualquier instante hay muy pocas de estas partículas reactivas de
vida breve. De las colisiones probables,
la que se realiza con una molécula de cloro no produce cambio neto; puede
ocurrir la reacción, pero sólo puede resultar en el intercambio de un átomo de cloro por otro:
![]()
La colisión de
un átomo de cloro con una molécula de metano es, a la vez, probable y
productiva. El cloro obtiene un átomo de hidrógeno con un solo electrón para
formar una molécula de cloruro de hidrógeno:
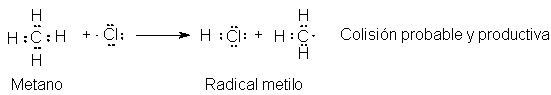
El grupo metilo queda con un electrón solitario,
no apareado; el átomo de carbono solamente tiene siete electrones en su capa de
valencia; ha sido consumido un radical libre, el átomo de cloro, y en su lugar
se ha formado otro, el radical metilo, CH3. Este es el paso (2) del
mecanismo.
Entonces, ¿qué
es lo que probablemente le sucederá al radical metilo? Al igual que el átomo de
cloro, es muy reactivo y por la misma razón: tiende a completar el octeto, a
perder energía formando un nuevo enlace. Nuevamente, las colisiones probables
son con moléculas de cloro o metano, y no con átomos de cloro o radicales
metilo, relativamente escasos. Sin embargo, el impacto con una molécula de
metano puede resultar, en el mejor de los casos, en el intercambio de un
radical metilo por otro:
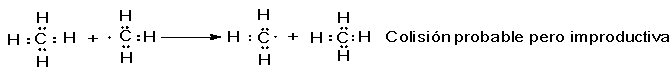
Por
consiguiente, la colisión importante es entre un radical metilo y una molécula
de cloro. El radical metilo toma un átomo de cloro con uno de los electrones enlazantes
para formar una molécula de cloruro de metilo:
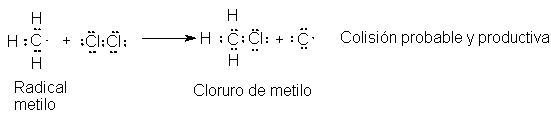
El otro producto es un átomo de cloro. Este es el
paso (3) del mecanismo.
También
aquí el consumo de una partícula reactiva va acompañado de la formación de
otra. El nuevo átomo de cloro ataca al metano
para generar un radical metilo que, a su vez, ataca a una molécula de cloro para dar un átomo de
éste, repitiéndose la secuencia una y otra vez. Cada paso no sólo genera una
nueva partícula reactiva, sino también una molécula de producto: cloruro de
metilo o cloruro de hidrógeno.
Sin embargo, este proceso no puede continuar
indefinidamente. Ya vimos que la unión de dos partículas de vida corta y
relativamente escasas es poco probable, pero sucede ocasionalmente, y cuando
sucede se detiene esta secuencia específica. Las partículas reactivas se
consumen, pero no se generan.
Está claro,
entonces, que el mecanismo explica los hechos (a), (b), (c), (d) y (e) de las
páginas 46 y 47; se necesita luz o calor
para romper la molécula de cloro y generar así los átomos de cloro iniciales;
una vez formado, cada átomo puede lograr la generación de muchas moléculas de
cloruro de metilo.
2.13 Reacciones en cadena
La cloración del metano es un ejemplo de reacción en cadena, una reacción qué comprende varios pasos,
cada uno de los cuales genera una sustancia reactiva que genera el paso
siguiente. Aunque las reacciones en cadena pueden variar mucho en sus
detalles, todas tienen ciertas
características en común.
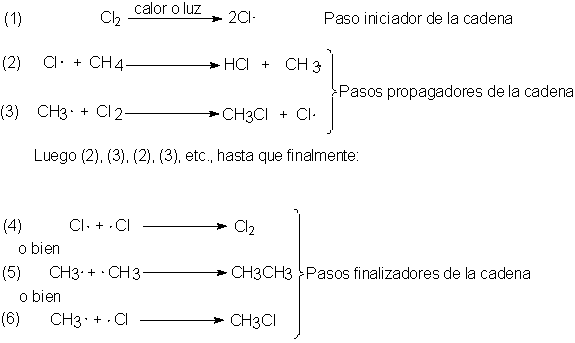
El primero de la cadena de reacciones es el paso iniciador, durante el cual se
absorbe energía y se genera una partícula reactiva; en la reacción considerada
se trata de la ruptura del cloro en átomos (paso 1).
Hay uno o más pasos
propagadores, cada uno de los cuales consume una partícula reactiva y
genera otra; en este caso se trata de la reacción de átomos de cloro con metano
(paso 2) y de radicales metilo con cloro (paso 3).
Por último, tenemos los pasos finalizadores, en los que se consumen partículas reactivas, pero no se generan; en la cloración del
metano éstos serían la unión de dos de las partículas reactivas o la captura de una de ellas por
las paredes del recipiente.
Dada una determinada serie de condiciones, se
forman alrededor de 10 000 moléculas de cloruro de metilo por cada cuanto de luz (fotón) absorbido. Cada
fotón rompe una molécula de cloro para generar dos átomos, cada uno de los
cuales inicia una cadena. En promedio, cada cadena consiste en 5000 repeticiones
del ciclo propagador antes de detenerse.
2.14 Inhibidores
Finalmente,
¿cómo explica este mecanismo el echo (f), que una pequeña cantidad de oxígeno
retarde la reacción durante un periodo de tiempo, el cual depende de la
cantidad de oxígeno para luego proceder normalmente?
Se cree que el oxígeno reacciona con un radical
metilo para formar un nuevo radical libre:
![]()
El radical CH3OO. Es mucho menos reactivo que el CH3., por lo
que poco puede hacer para continuar la cadena. Una molécula de oxígeno la interrumpe, al combinarse con
un radical metilo, evitando así la formación de miles de moléculas de cloruro
de metilo, lo que retardaría mucho la reacción. Una vez que todas las moléculas
de oxígeno presentes se han combinado con radicales metilo, la reacción
puede continuar a su velocidad normal.
Una
sustancia que retarda o detiene una
reacción, aun estado presente en cantidades pequeñas, se llama inhibidor. El tiempo durante el cual se
manifiesta la inhibición, y después del cual
la reacción procede normalmente, se denomina periodo de inhibición. La inhibición por una cantidad relativamente pequeña de material agregado
es bastante característica de las reacciones en cadena de todo tipo, y a menudo
es uno de los primeros indicios que nos
llevan a sospechar que estamos tratando con una reacción en cadena. Es
difícil imaginar de qué otro modo tan
pocas moléculas pueden impedir la reacción de tantas. (Nos encontraremos frecuentemente con el empleo de oxígeno para
inhibir reacciones de radicales libres.)
2.15 Calor de reacción
Al considerar la cloración del metano nos hemos
ocupado principalmente de las partículas implicadas moléculas y átomos y de los
cambios que sufren. Sin embargo, es también importante considerar, como para
cualquier reacción, los cambios energéticos involucrados, puesto que éstos son los que determinan en
gran medida su velocidad y si realmente se va a realizar.
Empleando los valores para las energías de
disociación homolítica de enlaces dados en la tabla 1.2 (Sec. 1.14), podemos
calcular los cambios energéticos que resultan para gran número de reacciones.
En la conversión del metano a cloruro de metilo, se rompen dos enlaces, CH3-H y CI-CI, con un
consumo de 104 + 58, o un total de 162 kcal/mol. Al mismo tiempo se forman dos
enlaces nuevos, CH3-CI y H-CI, con liberación de 84 + 103, o un total de 187 kcal/mol. El resultado es
la liberación de 25 kcal de calor por cada mol de metano convertido en cloruro
de metilo; así pues, ésta es una reacción
exotérmica. (Reconocemos que este cálculo no depende del conocimiento del
mecanismo de la reacción.)
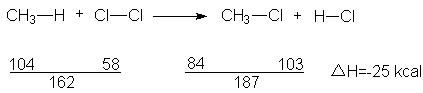
Cuando se libera energía, el contenido calórico
(entalpía), H, de las propias moléculas debe disminuir; en consecuencia, al cambio del contenido calórico, D H, se le
da un signo negativo. (En el caso de una reacción endotérmica, durante la cual
se absorbe calor, el aumento del contenido calórico de las moléculas se indica
por un D H positivo.)
El valor de -25 kcal recién calculado es el DH neto para la reacción global. Se obtiene un cuadro más útil del proceso
al considerar los DH de
los pasos individuales, que se calculan a continuación:
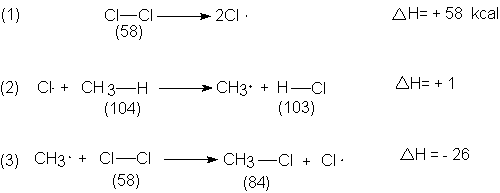
Es evidente por qué esta reacción sólo tiene lugar
a temperatura elevada (en ausencia de luz), a pesar de ser exotérmica. El paso
que inicia la cadena, sin el cual no puede realizarse el proceso, es muy endotérmico, por lo que sólo puede
ocurrir (con velocidad suficiente) a
temperatura alta. Una vez formados los dos átomos de cloro, los dos pasos que
propagan la cadena uno es ligeramente
endotérmico, y el otro, exotérmico se suceden sin dificultad muchas veces antes
de que la serie se interrumpa. La difícil ruptura del cloro es la barrera a
superar antes de que puedan funcionar las fáciles etapas siguientes:
Hasta este instante hemos supuesto que las
reacciones exotérmicas proceden sin dificultad, o sea, son razonablemente
rápidas a temperaturas ordinarias, mientras que las endotérmicas son lentas,
salvo a temperaturas elevadas. Esta relación supuesta entre DH y la
velocidad de reacción es una regla
empírica útil a falta de información adicional; sin
embargo, no es una relación necesaria,
y tiene muchas excepciones. Por esto, procederemos a estudiar otro concepto energético, el de energía de activación, relacionado más
precisamente con la velocidad de reacción.
2.16 Energía de activación
Para apreciar
lo que realmente sucede durante una reacción química, veamos un ejemplo
específico más en detalle, el ataque de átomos de cloro sobre el metano:
![]()
Esta reacción es relativamente sencilla: sucede en
fase gaseosa, y por eso no hay complicaciones por la presencia de algún
disolvente; involucra la interacción de
un solo átomos y la más simple de las moléculas orgánicas. Aun así podemos
aprender ciertos principios que son válidos para cualquier reacción.
¿Qué debe suceder para que esta reacción proceda?
En primer lugar, debe chocar un
átomo de cloro con una molécula de metano. Puesto que las fuerzas químicas son
de alcance muy corto, sólo puede formarse un enlace hidrógeno-cloro cuando los
átomos se encuentran en contacto íntimo.
Entonces, para que sea efectiva, la colisión debe suministrar cierta cantidad mínima de energía. La formación del enlace H-CI libera 103
kcal/mol; romper el enlace CH3-H requiere 104 kcal/mol.
Podríamos suponer que sólo se
necesitaría 1 kcal/mol adicional de
energía para que la reacción se realice,
no es así. Es evidente que la ruptura
y la formación de enlaces no
están perfectamente sincronizadas, por lo que no se dispone de todas la energía liberada por un proceso para el otro. Se ha demostrado
de modo experimental que deben suministrarse 4 kcal/mol adicionales para que la
reacción tenga lugar.
La
energía mínima que debe proporcionar una
colisión para que se produzca reacción se llama energía de activación, Eact,
cuya fuente es la energía cinética de las partículas en movimiento. La mayoría
de los impactos proporcionan menos que este mínimo, por lo que no prosperan y
las partículas originales simplemente rebotan. Sólo las colisiones
violentas entre partículas, de las
cuales una o ambas se mueven con velocidad desusada, son suficientemente
energéticas como para permitir la reacción. En este ejemplo, a 275ºC sólo una
colisión de cada 40 es suficientemente energética.
Por último, además de ser suficientemente
energéticos, los choques deben producirse
entre partículas orientadas
en forma correcta. En el instante de la colisión, la molécula de metano debe
tener una orientación tal que presente un átomo de hidrógeno a la fuerza
plena del impacto. En nuestro ejemplo,
sólo alrededor de un choque de cada ocho tiene orientación apropiada.
En general, una
reacción química requiere colisiones de
energía suficiente (Eact) y
de orientación apropiada. Existe una energía de activación para casi toda
reacción que implica ruptura de enlaces,
incluso para reacciones exotérmicas, en las que la formación de
enlaces libera más energía que la
consumida en la ruptura.
El ataque de átomos de bromo sobre el metano es
mucho más endotérmico, con un DH de + 16
kcal.
![]()
La ruptura del enlace CH3 - H requiere,
como en el caso anterior, 104 kcal/mol, de las que la formación de la unión
H-Br sólo proporciona 88 kcal. Es evidente que aunque se dispusiera totalmente
de estas 88 kcal, la colisión debería
suplir por lo menos 16 kcal adicionales. En otras palabras, la Eact de
una reacción endotérmica debe ser al menos de la magnitud del DH. Como generalmente es cierto, la Eact de esta reacción
particular (18 kcal) es, de hecho, algo mayor que el DH.
2.17 Avance de la reacción: cambios de energía
Las relaciones de energía pueden apreciarse más
claramente en diagramas como los de las figuras 2.3 y 2.4. El avance de la
reacción se representa por un movimiento
horizontal desde los reactivos, a la izquierda, hacia los productos, a la
derecha. La energía potencial ( es decir, toda energía, excepto la cinética) en
cualquier etapa de la reacción esta indicada por la altura de la curva.
Sigamos el curso de la reacción en la figura 2.3.
Partimos de un vale de energía potencial con una molécula de metano y un átomo de cloro. Estas partículas se
mueven, por lo que poseen energía cinética, además de la energía potencial
indicada. La cantidad exacta de energía cinética varía con cada par de
partículas, puesto que algunas se mueven más velozmente que otras. Al
chocar, la energía cinética se convierte
en energía potencial. Con el aumento de la energía potencial comienza la
reacción y empezamos a remontar la curva energética. Si se convierte suficiente
energía cinética, alcanzamos la cima y empezamos a descender hacia la derecha.
Durante el descenso se convierte energía potencial
en cinética hasta que alcanzamos el
nivel de los productos, que contienen algo más de energía potencial que
los reactivos, y así nos encontramos en un valle ligeramente más elevado que el que habíamos dejado. Con
este incremento neto de energía potencial debe haber una disminución correspondiente de energía cinética. Las partículas nuevas se
distancian y, puesto que se mueven más
lentamente que las partículas de las
cuales se formaron, observemos un descenso de la temperatura: se absorbe calor
del entorno.
En la reacción del bromo. Indicada en la figura
2.4, ascendemos una curva mucho más alta y terminamos en un valle también más
elevado. El aumento de energía potencial y la disminución correspondiente de
energía cinética es mucho más acentuado que en la reacción del cloro: se
absorbe más calor del entorno.
Una
reacción exotérmica sigue un curso muy similar. (Como ejemplo, obsérvese
la reacción inversa del bromo, es decir, lea de derecha a izquierda en la Fig.
2.4.) En este caso, sin embargo, los productos con menos energía potencial que los reactivos terminan en un valle más
bajo que el que habíamos dejado. Puesto que esta vez las partículas nuevas contienen más energía cinética que las de
origen, por lo que se mueven con mayor velocidad, observamos un aumento de la
temperatura; se entrega calor al
entorno.
En toda reacción se producen muchas colisiones que
no liberan suficiente energías para
alcanzar la cumbre de la colina; estas colisiones son estériles, por lo
que nuevamente terminamos en el valle primitivo. Muchas colisiones proporcionan
energía suficiente, pero suceden con las
moléculas mal orientadas; esto nos permite subir una colina energética,
pero nos encontraremos fuera de ruta: podemos subir mucho sin encontrar el paso
que permite el acceso al valle siguiente.
La
diferencia de nivel entre ambos valles es, desde luego, DH y la
diferencia de nivel entre el valle de los reactivos y la cima es la Eact. Sólo nos interesan estas diferencias, no la
altura absoluta de una etapa de la reacción; ni siquiera nos interesan los
niveles relativos de los valles de los reactivos en las reacciones del cloro y
bromo: solamente necesitamos saber que
en la reacción del cloro remontamos una colina de 4 kcal de altura para
terminar en un valle 1 kcal más elevado que nuestro punto de partida, y que en
la reacción del bromo, subimos una
altura de 18 kcal para finalizar en un valle 16 kcal más alto que el inicial.
Veremos que la altura de la curva, Eact,
es la que determina la velocidad de la
reacción, y no la diferencia de nivel, D H, de
los dos valles. Al pesar a un valle más bajo, la colina puede ser muy alta,
pero podría ser baja e incluso
inexistente. Sin embargo, al remontarnos hacia un valle más elevado, la colina no puede ser más baja
que el valle al cual vamos, es decir, en una reacción endotérmica, Eact
debe ser, por lo menos, igual a DH.
Un diagrama de energía del tipo ilustrado en las
figuras 2.3 y 2.4 es particularmente útil, porque no sólo describe la reacción
que estamos considerando, sino también la inversa: por ejemplo,
observemos la figura 2.3 de derecha a izquierda. Vemos que la reacción
tiene una energía de activación de 3 kcal, puesto que en este caso remontamos
la curva desde el valle superior; ésta
es, claro está una reacción exotérmica, con un
DH de -1 kcal.
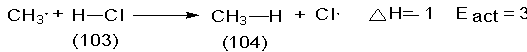
Del mismo modo, podemos deducir de la figura 2.4 que la reacción
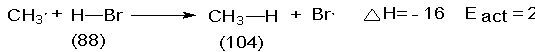
tiene una energía de activación de 2 kcal, siendo
exotérmica, con un DH de -16 kcal.
(Observemos que
estas dos últimas reacciones tienen energía de activación, a pesar de ser
exotérmicas.)
Reacciones como la
ruptura del cloro en átomos pertenecen a una categoría especial:

se rompe un
enlace, pero no se forman otros. La reacción inversa, la unión de dos átomos de
cloro, no implica ruptura de enlaces, y es de esperar que proceda con
facilidad:

de hecho, sin energía de activación. Esto se
considera válido para toda reacción que involucre la unión de dos radicales
libres.
Si no hay
obstáculo que vencer al pasar de átomos a una molécula de cloro, sino
sólo descender una pendiente, la ruptura de una molécula de cloro debe
simplemente implicar el ascenso de una
pendiente, como ilustra la figura 2.5. Entonces, la Eact para la
ruptura de esta molécula debe ser igual a DH, o 58
kcal. Esta igualdad de Eact y DH se
cree generalmente válida para reacciones de disociación de moléculas en
radicales.
2.18 Velocidad de reacción
Una reacción química es el resultado de colisiones
suficientemente energéticas y adecuadamente orientadas. La velocidad de reacción,
en consecuencia, debe ser la velocidad a la que suceden estos impactos
efectivos, el número de colisiones efectivas que ocurren, digamos durante cada
segundo dentro de cada litro de espacio
de reacción. En consecuencia, podemos expresar la velocidad de reacción como el
producto de tres factores. (El número que expresa la probabilidad de que una
colisión tenga la orientación adecuada se suele denominar factor de probabilidad.) Todo lo que afecte a cualquiera de estos
factores afectará a la velocidad de reacción.

La frecuencia
de colisiones depende de: (lo aglomeradas que están las partículas, es
decir, concentración o presión; (b) lo grandes que son, y (c) lo rápido que se mueven, lo que, a su
vez, depende de su peso y de la temperatura.
Podemos variar
la concentración y la temperatura y, en consecuencia, la velocidad. Estamos
familiarizados con el hecho de que un aumento en la concentración produce un
aumento de la velocidad: esto sucede, desde luego, porque aumenta la frecuencia
de las colisiones. Un aumento de la
temperatura también la incrementa; como veremos, también aumenta el factor energético, siendo
este último efecto tan grande que, en comparación, la influencia de la
temperatura sobre la frecuencia de impactos carece de importancia.
El tamaño y peso de las partículas son
característicos de cada reacción y no pueden
cambiarse; aunque varían ampliamente de una reacción a otra, esta
variación no afecta demasiado a la frecuencia de las colisiones. A cierta
temperatura, un peso mayor hace moverse más lentamente a una partícula, con lo
que tiende a disminuir la frecuencia de las colisiones; sin embargo, una
partícula más pesada es, por lo general, una partícula más grande y el mayor tamaño tiende a aumentar la
frecuencia de los choques. De este modo, estos dos factores tienden a anularse.
El factor
de probabilidad depende de la geometría de las partículas y del tipo de
reacción que se esté realizando. Para reacciones muy relacionadas, no varía
grandemente.
La energía cinética de las moléculas en movimiento
no es la única fuente de la energía necesaria para reaccionar; por ejemplo,
puede proporcionarse energía de las vibraciones de los diversos átomos de
la molécula de modo que el factor de
probabilidad no sólo tiene que ver con qué átomos de la molécula sufren colisión, sino también con la
ubicación de los demás átomos en ella, en el instante del choque.
El factor más importante para determinar la
velocidad es, con mucha diferencia, el factor energético: la fracción de colisiones
suficientemente energéticas. Este factor depende de la temperatura, que podemos
controlar, y de la energía de activación característica de cada reacción.
A una temperatura dada, las moléculas de un
compuesto determinado tienen una velocidad
promedio y, en consecuencia, una energía cinética media que es
característica de ese sistema; de hecho, la temperatura es una medida de esta
energía cinética media. Sin embargo, no todas las moléculas individuales se
mueven con la misma velocidad: unas son
más rápidas y otras más lentas que el promedio. La distribución de
velocidades se ilustra en la figura 2.6 con la conocida curva campaniforme que
describe la distribución entre
individuos de una gran diversidad de cualidades, como altura, inteligencia,
ingresos e incluso expectativa de vida. El número de moléculas con una energía
cinética determinada es mayor para una energía próxima a la medida y decrece a
medida que ésta se hace mayor o menor que el promedio.
La distribución de energías de choque, como era de
esperar, se ilustra con una curva similar (Fig. 2.7). Indicaremos las
colisiones de una energía determinada, Eact, por medio de una línea
vertical; el número de colisiones con energía igual o mayor que Eact
está indicado
por el área sombreada bajo la curva, a la derecha de la línea
vertical. La fracción del número total de choques que tienen esta energía mínima, Eact, es la
fracción del área total que está sombreada. Resulta evidente que cuanto mayor
sea el valor de Eact, tanto
menor es la fracción de colisiones que tienen esa energía.
La relación exacta entre energía de activación y
fragmentación de colisiones con esa energía es:
e-Eact/RT
= fracción de colisiones con energía mayor que Eact,
donde
Eact = energía de activación en cal (
no kcal)
e =
2.718 ( base de logaritmos naturales)
R= 1.986
(constante de los gases)
T=
temperatura absoluta.
Empleando P para el factor de probabilidad y Z
para la frecuencia de colisiones, obtenemos la ecuación de velocidad:
velocidad
PZe-Eact/RT
Esta relación exponencial es importante porque
indica que una diferencia pequeña en Eact
tiene un efecto considerable sobre la fracción de colisiones suficientemente
energéticas y, en consecuencia, sobre la velocidad de reacción. A 275ºC, por
ejemplo, de cada 1 000 000 de choques, 10 000 suministran energía suficiente si
Eact=5 kcal; 100 la proporcionan si
Eact=10 kcal, y solamente lo hace una si Eact=15
kcal. Esto significa que (permaneciendo iguales todos los demás factores) una
reacción con Eact=5 kcal, procederá 100 veces más rápidamente que
una con Eact=10 kcal, y 10 000 veces más velozmente que una con Eact=15
kcal.
Hasta aquí hemos considerado un sistema mantenido
a cierta temperatura. Un incremento de
ésta aumenta, claro está, la energía cinética y las velocidades promedio, con
lo que la curva toda se desplaza a la derecha, como se indica en la figura 2.8.
Por tanto, para una energía de activación determinada, un aumento de la
temperatura incrementa la fracción de colisiones suficientemente energéticas y
en consecuencia, como bien sabemos, aumenta la velocidad.
Nuevamente, la relación exponencial conduce a un gran cambio de velocidad, esta vez por
una pequeña variación de la temperatura. Así, por ejemplo, un aumento de 250ºC
a 300ºC, que sólo representa un
incremento del 10% en temperatura absoluta, aumenta la velocidad en un 50% si Eact=5
kcal; la dobla, si Eact=10
kcal, y la triplica, si Eact = 15 kcal. Como ilustra este ejemplo,
cuanto mayor sea Eact, mayor será el efecto de un cambio de
temperatura determinado, lo que se desprende de la relación e-Eact/RT.
De hecho, esta relación entre velocidad y temperatura es la que permite
determinar la Eact de una reacción: se mide la
velocidad a diferentes temperaturas, y con los resultados obtenidos se
calcula Eact.
Hemos examinado los factores que determinan la
velocidad de una reacción, y podemos emplear de muchas maneras lo estudiado.
Por ejemplo, para acelerar una reacción,
sabemos que podemos elevar la temperatura o aumentar la concentración de
reactivos, e incluso disminuir la Eact (Por medios que veremos más
adelante).
De interés inmediato, sin embargo, es la cuestión
de las reactividades relativas. En consecuencia, veamos cómo nuestro
conocimiento de las velocidades de reacción nos puede ayudar a explicar el
hecho de que una reacción procede más velozmente que otra, a pesar de que las condiciones sean idénticas para
ambas.
2.19 Velocidad relativa de reacción
Hemos visto que la velocidad de una reacción puede
expresarse por el producto de tres factores:
velocidad
= frecuencia de colisión X factor
energía X factor
probabilidad
Dos reacciones pueden proceder a una velocidad
distinta debido a diferencias en uno de estos factores o en todos. Para
explicar una diferencia en la velocidad debemos ver primero en cuál de estos
factores está la diferencia.
Como ejemplo, comparemos las reactividades de átomos de cloro y bromo
hacia el metano, es decir, comparemos la velocidad de cada reacción en
condiciones iguales:
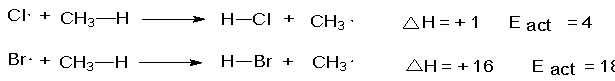
Puesto que la temperatura y la concentración deben
ser iguales para ambas reacciones, si
hemos de compararlas en igualdad de condiciones, cualquier diferencia en la frecuencia de colisión tiene que deberse al tamaño o
peso de las partículas. Un átomo de bromo es más pesado y más grande que uno de
cloro; vimos que los efectos de estas dos propiedades tienden a compensarse; de
hecho, las frecuencias de colisión sólo difieren en un pequeño porcentaje. Generalmente, es cierto, para una
misma temperatura y concentración, dos reacciones íntimamente relacionadas
apenas difieren en frecuencia de
colisión; por consiguiente, ésta no puede ser la causa de una gran diferencia
en la reactividad.
Se conoce muy poco la naturaleza del factor
probabilidad, pero como ambas reacciones son muy similares, es de suponer
que tengan factores parecidos, lo que se
confirma experimentalmente: sólo alrededor
de una colisión de cada ocho con metano tiene una orientación propicia para
reaccionar, trátese de átomos de cloro o de bromo. En general, cuando se trata de reacciones muy semejantes, podemos suponer que no es probable que una gran
diferencia de reactividad se deba a factores de probabilidad muy distintos.
Nos queda por considerar el factor energía. A una temperatura dada, la fracción de choques con
suficiente energía para reaccionar depende de su magnitud, o sea, depende de su
Eact. En nuestro ejemplo, Eact es de 4 kcal para la
reacción del cloro, y de 18 kcal para la del
bromo. Hemos visto que una diferencia de este orden es la causa de una diferencia enorme en el factor energético,
y por tanto en la velocidad. A 275ºC, de cada 15 millones de colisiones, 375 000 son suficientemente
energéticas cuando implican átomos de
cloro, y solamente una con átomos de bromo. En consecuencia, por la sola
diferencia en Eact, los átomos de cloro son 375 000 veces más
reactivos con el metano que los de bromo.
A medida que nos encontramos una y otra vez con
diferencias en la reactividad, generalmente las atribuiremos a diferentes Eact;
en muchos casos, podremos explicarlas por las
diferencias de estructura molecular. Debe
entenderse que sólo se justifica proceder
así cuando las reacciones que se
comparan están tan íntimamente
relacionadas que las diferencias en la frecuencia de colisión y en el factor de
probabilidad son comparativamente insignificantes.
2.20 Reactividades relativas de los halógenos con el
metano
Con estos antecedentes, retornemos a la reacción
entre el metano y los diferentes halógenos, y veamos si podemos explicar el
orden de reactividad indicado antes: F2 > CI2 > Br2 > I2, y en particular el hecho de
que el yodo no reaccione.
La tabla de energías de disociación de enlaces (Tabla 1.2, Sec.
1.14) nos permite calcular para cada halógeno el DH de
cada uno de los tres pasos de la halogenación. Puesto que Eact ha
sido determinada sólo para algunas de estas reacciones, veamos qué conclusiones
provisionales podemos alcanzar empleando solamente los DH.
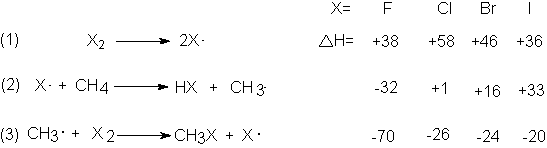
Puesto que el paso (1) solamente implica la
disociación de moléculas en átomos, podemos suponer confiadamente que en este
caso DH es igual a Eact (Sec. 2.17 y Fig. 2.5). El cloro tiene la Eact
más elevada, por lo que debería ser el más lento en disociarse; en cambio, el yodo tiene la menor Eact,
debiendo ser, en consecuencia, el de disociación más rápida. Sin embargo, esto
no concuerda con el orden de reactividad observado, por lo que la disociación
del halógeno en átomos no puede ser la
etapa que determine las reactividades conocidas, salvo quizá para el flúor.
El paso (3), el ataque de radicales metilo al
halógeno, es exotérmico para los cuatro halógenos, siendo el DH casi
igual para el cloro, el bromo y el yodo. La Eact podría ser muy baja para estas
reacciones, y de hecho parece ser así: probablemente sólo una fracción de una
kilocaloría. Incluso se ha observado que el yodo reacciona con facilidad con
radicales metilo generados de otro modo, por ejemplo, por calentamiento de
tretrametilplomo. De hecho, a veces se
emplea yodo como <<trampa>> para radicales libres en estudios de
mecanismos de reacción. Por
consiguiente, tampoco la tercera etapa puede ser la causa de las reactividades relativas.
Esto nos deja
con el paso (2), la separación de hidrógeno del metano por medio de un
átomo de halógeno. Para éste, observamos un intervalo amplio de valores de DH que
va desde la reacción muy exotérmica con
flúor hasta la fuertemente endotérmica
con el yodo. La reacción endotérmica del
átomo de bromo debe tener una Eact de al menos 16 kcal; como hemos visto es de 18 kcal. La
reacción ligeramente endotérmica del átomo de cloro podría tener una Eact muy baja, y es
efectivamente de 4 kcal. Luego, a una temperatura dada, la fracción de colisiones suficientemente
energéticas es mucho mayor para los átomos de cloro y metano que para los de bromo y metano; para ser específicos, a
275ºC, esta fracción es de alrededor de 1 en 40 para el cloro y solamente de 1
en 15 millones para el bromo.
En promedio, un átomo de bromo choca con muchas
moléculas de metano antes de lograr la separación de hidrógeno, mientras que uno de cloro choca
relativamente con pocas. Durante su búsqueda
más prolongada de la molécula de
metano apropiada, un átomo de bromo
tiene mayor probabilidad de encontrar otra partícula escasa un segundo átomo de
bromo o un radical metilo o de ser capturado por las paredes del recipiente;
por consiguiente, las cadenas deberían ser mucho más cortas que en la
cloración, lo que de hecho se ha determinado experimentalmente: mientras que
para la cloración la cadena es de varios
miles de pasos, para la bromación es de menos de 100. A pesar de que los
átomos de bromo se generan más rápido que los de cloro por la menor Eact
del paso (1), la bromación global es más lenta que la cloración, debido a la
menor extensión de la cadena.
Para la reacción endotérmica de un átomo de yodo
con el metano, la Eact no puede ser
inferior a 33 kcal, probablemente es algo mayor. A un para este mínimo
de 33 kcal, un átomo de yodo debe chocar
con un número enorme de moléculas de metano (1013, ó 10 billones, a 275ºC), antes que se dé la probabilidad de
reacción. Virtualmente ningún átomo de
yodo dura tanto, sino que se recombina para formar moléculas de yodo, por lo que la reacción procede a velocidad
imperceptible. Es fácil egnerar átomos de yodo; lo que impide la yodación es su incapacidad de separar
hidrógeno del metano.
No podemos
predecir la Eact para el ataque muy exotérmico del flúor al metano, pero
en ningún caso puede ser mayor que para el ataque de los átomos de cloro; de
hecho, parece ser menor (alrededor de 1 kcal), lo que permite cadenas aún más largas. Debido a la sorprendente
debilidad del enlace flúor-flúor, los átomos correspondientes deben generarse
más rápidamente que los del cloro, por
lo que no sólo deben ser más extensas
las cadenas, con un DH de 102 kcal, siendo una de las causas de su difícil control, la
dificultad de eliminar el calor generado.
En consecuencia, de las dos etapas propagadoras de la
cadena, el paso (2) es más difícil que
el (3) (véase Fig. 2.9). Una vez formados, los radicales metilo reaccionan fácilmente
con cualquiera de los halógenos; lo que limita la velocidad de la reacción
global es la rapidez de la formación de radicales metilo. La fluoración es
rápida porque los átomos de flúor extraen
rápidamente hidrógeno del metano; su Eact es tan sólo de 1
kcal. La yodación no tiene lugar porque es virtualmente imposible para los
átomos de yodo extraer hidrógeno del metano; su Eact es superior a
33 kcal.
Podemos apreciar que los valores de Eact para el
paso (2) son paralelos a los de DH.
Puesto que en cada caso se rompe el mismo enlace
CH3-H, las diferencias de DH reflejan diferencias en energías de disociación entre los diversos
enlaces hidrógeno-halógeno. En definitiva, parece que la reactividad de un halógeno hacia el metano depende de la
fuerza del enlace que dicho halógeno
forma con el hidrógeno.
Hay un aspecto adicional que requiere aclaración:
hemos dicho que una Eact de 33 kcal es demasiado grande para que la reacción entre el metano y átomos
de yodo se produzca a velocidad apreciable; sin embargo, el paso inicial de
cada una de estas halogenaciones requiere una Eact aún mayor. La
diferencia es la siguiente: puesto que la halogenación es una reacción en
cadena, la disociación de cada molécula de halógeno acaba generando muchas
moléculas de halogenuro de metilo; en consecuencia, aunque la disociación sea
muy lenta, la reacción global puede ser rápida. Sin embargo, el ataque de los
átomos de yodo al metano es un paso propagador de cadena,
y si es
lento, toda la reacción debe ser lenta; en estas circunstancias, los pasos que
la terminan (por ejemplo, la unión de dos átomos de yodo) pasan a ser tan
importante que, de hecho, no hay cadena.
2.21 Mecanismo alternativo para la halogenación
En la sección anterior nos ocupamos de las
reactividades relativas de los diversos halógenos hacia el metano. En el
siguiente capítulo modificaremos nuestro punto de vista y estudiaremos las
reactividades relativas de varios alcanos -o de diferentes posiciones en un
mismo calcano con un halógeno determinado. Todo esto ayuda a construir una
parte importante de nuestro estudio de la química orgánica: la manera en que las variaciones
estructurales modifican la reactividad. Hay, no obstante, un aspecto aún más fundamental que debe ser
considerado: cómo, en primer lugar, una estructura particular conduce a
un tipo particular de reacción. La
cuestión no está en que un halógeno o un alcano reaccione más rápido o más lento que otro, sino en que
cualquier halógeno y cualquier alcano reaccionen juntos del modo que lo hacen.
Para dar
respuesta a esta cuestión, tomemos como ejemplo la cloración del metano y
examinémosla cuidadosamente. Las etapas que propagan la cadena en nuestro
mecanismo son (2a) y (3a).
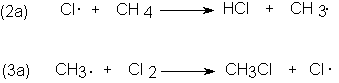
Consideremos, en cambio, la secuencia (2b) y (3b),
que representa un mecanismo alternativo.
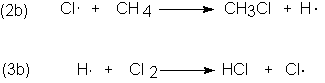
Según puede apreciarse, este mecanismo es
ciertamente digno de ser considerado; de hecho, hasta 1940 resultaba tan
evidente como lo es aceptado actualmente. Sin embargo, la cloración no sigue este
mecanismo alternativo, y en la sección 4.28 presentaremos pruebas directas en
su contra.
Por tanto, nuestra pregunta pasa a ser siguiente:
¿Por qué la cloración sigue los pasos (2a) y (3a), y no los (2b) y (3b)? La
clave del problema está en el paso (2). En este punto se dividen las dos sendas
de reacción: lo que sucede en (2) determina el curso entero de la reacción. Si
ocurriese (2b), seguiría inevitablemente (3b); (3b) es una reacción conocida que sucede con facilidad en
un sistema diferente. Pero (2b) no sucede.
Hemos
estrechado así aún más nuestra
averiguación. Preguntamos ahora: ¿Por qué sucede (2a) en lugar de (2b)? En ambas reacciones, el
átomo de cloro ataca a una molécula de metano. Puede unirse a un hidrógeno y
expulsar un radical metilo, o unirse a un carbono y expulsar un átomo de
hidrógeno. Por tanto, hay competencia entre ambas reacciones, y gana la más
rápida. Si predomina el paso (2a), sólo puede significar que (2a) es más rápida
que (2b).
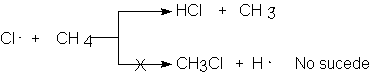
¿Cómo se explica esto? Idealmente, nos interesaría
conocer Eact para las reacciones en competencia, pero ciertamente es
imposible medir Eact para (2b), puesto que esta reacción no se lleva
a cabo, Por tanto, veamos lo que podemos hacer empleando valores de DH, como
hicimos en la sección 2.20: podemos calcular éstos para reacciones reales o
imaginarias utilizando las energías de disociación homolítica de enlaces de la
tabla 1.2. Para (2a), DH es de + 1 kcal; en consecuencia,
la Eact podría ser tan pequeña como este valor y, como ya sabemos,
es realmente de 4 kcal.
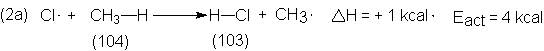
Para (2b), DH es de
+ 20 kcal; por tanto, Eact debe ser por lo menos de 20 kcal; y quizá
sea mucho mayor.
![]()
La fracción de colisiones que proporcionan 4 kcal
o más es muchísimo mayor que la que
proporciona 20 kcal: a 275ºC, por ejemplo, es 2.5 millones de veces
mayor. Sólo teniendo en cuenta esta estimación mínima de la diferencia en Eact,
podemos observar que (2a) debe proceder con una rapidez tan superior a la de
(2b), que, de hecho, (2a) es la única reacción que se realiza.
La cuestión no es que 20 kcal sea en si una
barrera demasiado alta para impedir la reacción; al fin y al cabo, el ataque
del Br al metano tiene una Eact de 18 kcal, y ocurre. La cuestión
aquí reside en que una reacción con Eact de 20 kcal no puede competir
con éxito con otra cuya Eact sea tan sólo de 4 kcal. Cuando un átomo
de cloro choca con una molécula de
metano, esta colisión tiene una probabilidad abrumadoramente mayor de
proporcionar energía suficiente a (2a) que a (2b); por eso tiene lugar (2a).
Finalmente, veamos que característica estructural
es la que hace más fácil (2a). Ambas reacciones implican la ruptura de un enlace carbono-hidrógeno. La diferencia
está en cuál es el enlace que se formará: cloro-hidrógeno o cloro carbono. La
ruptura del enlace carbono-hidrógeno
requiere 104 kcal/mol - una cantidad considerable de energía-. Una
fracción pequeña de ésta está
proporcionada por colisiones, pero la mayor parte proviene de la formación
concertada de otro enlace: hidrógeno-cloro en el caso (2a), carbono-cloro, en el de (2b). El primero de éstos es fuerte (103
kcal), y su formación puede proporcionar casi toda la energía necesaria. El
enlace cloro-metilo, en cambio, es más
débil (sólo 84 kcal) y aunque
toda esa energía estuviese disponible para ayudar a romper la unión
carbono-hidrógeno, siempre habría que obtener 20 kcal más mediante colisiones.
El transcurso de esta reacción queda determinado, por tanto, por el hecho de
que el enlace cloro-hidrógeno es más fuerte que el cloro-metilo.
El examen de las energías de disociación de
enlaces de la tabla 1.2 nos indica que lo recién descrito es parte de una
tendencia: cada halógeno forma una unión
más fuerte con hidrógeno que con carbono no sólo con el carbono del metano,
sino también con el de otros alcanos-. El resultado es que, cualquiera que sea
el halógeno y el alcano, la halogenación
sigue un mecanismo análogo a (2a) y (3a), y no a (2b) y (3b).
Nuevamente
hemos tratado con velocidades relativas de reacciones, esta vez para
determinar el aspecto más fundamental del comportamiento químico: el tipo de reacción que se lleva a cabo.
Cuando se mezclan tipos diferentes de
moléculas, habrá, en principio, más de una forma de reacción entre ellas. Habrá
competencia entre caminos diferentes de reacción muy a menudo, como veremos, la
competencia será más cerrada que la
recién utilizada como ejemplo-. Y, en
todo respecto, lo que las moléculas realmente hacen, es lo que resulta más simple para ellas. A medida que encontramos
tales casos de competencia, trataremos de dilucidar los factores que tiendes a
favorecer un camino u otro; incluso
trataremos de ver qué podemos hacer para que la senda que preferimos sea la más
fácil a seguir para la reacción.
2.22 Estructura del radical metilo. Hibridación sp2
Hemos empleado buena parte de este capítulo en
estudiar la formación y las reacciones del radical libre metilo, CH3.
¿Cómo es esta molécula? ¿Cuál es su forma?¿Cómo se contribuyen sus electrones
y, en particular, dónde se encuentra el
electrón impar?
Estas son preguntas importantes, porque las
respuestas correspondientes son válidas no sólo para este radical sencillo,
sino para cualquier radical libre que
encontremos, cualquiera que sea su complejidad. Naturalmente, la forma
depende de la química tridimensional -la
estereoquímica de los radicales libres. La
ubicación de su electrón impar está íntimamente relacionada con la
estabilización de radicales libres por grupos sustituyentes.
Tal como procedimos al <<construir>> el
metano (Sec. 1.11), comenzamos con la configuración electrónica del carbono.

Y, para disponer de más de dos electrones no
apareados para enlaces, promovamos un electrón 2s al orbita 2p vacío:

Aquí el carbono se encuentra unido a otros tres
átomos, como lo está el boro en el trifluoruro de boro (Sec. 1.10). La
hibridación del orbital 2s con dos de los orbitales p suministra los orbitales
necesarios: tres sp2 fuertemente direccionales que, como vimos
antes, se encuentran en un plano que incluye el núcleo de carbono y dirigidos hacia
los vértices de un triángulo equilátero.
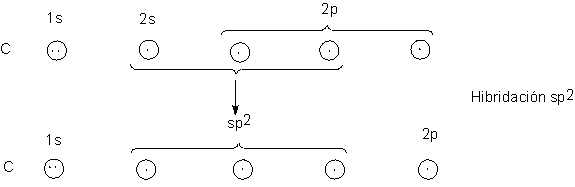
Si cambiamos el carbono y los tres hidrógenos de
un radical metilo de modo que el solapamiento de orbitales sea máximo,
obtenemos la estructura de la figura 2.10a. Es plana, con el átomo de carbono
en el centro de un triángulo y los tres hidrógenos en los vértices.
Todos los
ángulos de enlace son de 120º.
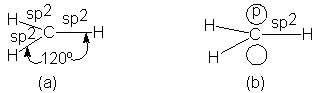
Fig. 2.10
Radical metilo. (a) Se indican solamente enlaces o. (b) Electrón impar en
orbital-p sobre y bajo el plano de los enlaces o.
Ahora, ¿dónde se encuentra el electrón impar? Al
formar los orbitales sp2, el carbono sólo empleó dos de sus tres
orbitales p. El restante consiste en dos lóbulos iguales, uno situado sobre el
plano y el otro debajo de los tres orbitales sp2 (Fig. 2.10b); éste
es ocupado por el electrón impar.
Esta no es la única configuración electrónica
concebible para el radical metilo: un tratamiento alternativo conduciría a una molécula piramidal como la del
amoniaco, excepto que el cuarto orbital sp3 contiene el electrón
impar, en vez de un par de electrones (Sec. 1.12). Los cálculos de mecánica
cuántica no ofrecen una decisión clara entre ambas configuraciones, pero los
estudios espectroscópicos indican que el radical metilo realmente es plano, o
casi plano. El carbono es trigonal, o bien se aproxima a serlo; el electrón
impar ocupa un orbital p o, por lo
menos, uno con mucho carácter p.
Compare la forma de tres moléculas, cuyo átomo
central está unido a otros tres: (a) trifluoruro de boro, sin electrones no compartidos, trigonal; (b)
amoniaco, con un par no compartido,
tetraédrico, y (c) el radical metilo, con un solo electrón no apareado, trigonal, o intermedio entre trigonal y
tetraédrico.
Hay pruebas estereoquímicas (por ejemplo, Sec.
4.28) de que la mayoría de los radicales libres son planas o bien, si son
piramidales, sufren inversión rápida,
tal como la molécula de amoniaco (Sec. 1.12).
2.23 Estado de transición
El concepto de Eact debe ser nuestra
clave para la comprensión de la reactividad química, pero para hacerla útil necesitamos un
concepto adicional: el estado de
transición.
Probablemente una reacción química es un proceso
continuo que implica una transición gradual de reactivos a productos. Sin
embargo, ha resultado útil considerar la disposición de los átomos en una etapa
intermediaria de la reacción, como si se tratara de una molécula real. Esta
estructura intermedia se denomina estado
de transición; su contenido de energía corresponde al máximo de la curva de
energía (Fig. 2.11).
Tal como DH es la diferencia en contenido
energético entre reactivos y productos, Eact es la diferencia en contenido de energía entre reactivos y estado de
transición.
El concepto de estado de transición es útil por
esta razón: podemos analizar su estructura como si se tratara de una molécula e
intentar estimar su estabilidad. Todo
factor que estabiliza el estado de
transición en relación con los reactivos tiende a disminuir la energía de activación;
es decir, todo factor que rebaja la cima de la colina energética más que el
valle de los reactivos reduce la altura
neta que debe vencer durante la reacción. En este libro, la estabilidad del
estado de transición será la base - explícita o implícita - de, prácticamente,
todo estudio de la reactividad.
Pero el estado de transición es sólo una
disposición pasajera de átomos que, por su naturaleza intrínseca encontrándose
en la cima de la colina energética -, no puede ser aislado y examinado. ¿Cómo
podemos llegar a saber algo acerca de su estructura? Pues bien, tomemos como ejemplo
el estado de transición para la separación de hidrógeno del metano por un átomo
de halógeno y vemos a qué nos conduce reflexionar un poco.
Para comenzar,
con seguridad podemos decir lo siguiente: el enlace carbono-hidrógeno se estira,
pero no se rompe del todo, mientras que la unión hidrógeno-halógeno ha
comenzado a formarse, aunque sin completarse. Esta condición puede
representarse por
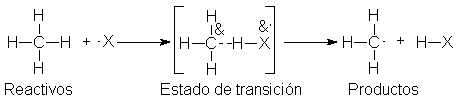
en donde las
líneas de puntos indican enlaces parcialmente rotos o formados.
Ahora, ¿qué podemos decir acerca de la forma del
grupo metilo en este estado de transición? En el reactivo, en el cual el grupo
metilo está unido al hidrógeno, el carbono es
tetraédrico (con hibridación sp3); en el producto, en el que el metilo ha perdido el hidrógeno, el
carbono es trigonal (hibridación sp2). En el estado de transición,
con el enlace carbono hidrógeno
parcialmente roto, la hibridación del carbono es intermedia entre sp3
y sp2; el metilo se ha
aplanado parcialmente, aunque no del todo; los ángulos de enlace son mayores
que 109.5, pero menores que 120º.
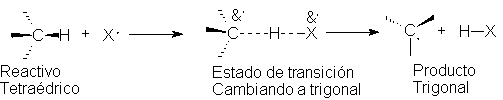
Por último, ¿dónde se encuentra el electrón impar?
Se hallan sobre el cloro en los reactivos, sobre el metilo en los productos y,
compartido entre ambos, en el estado de transición. (La parte correspondiente a
cada átomo se representa por &-.) El grupo metilo soporta parcialmente el electrón impar que
tendrá en el producto, con lo que ha adquirido,
en la proporción correspondiente, algo de las características del
radical libre que llegará a ser.
De este modo muy directo, hemos trazado una
descripción del estado de transición, que expone la ruptura y al formación de
enlaces, la disposición espacial de los átomos y la distribución de los
electrones.
(Este estado de transición específico es intermediario
entre reactivos y productos, no sólo en la secuencia del tiepo, sino también
estructuralmente. No todos los
estados de transición son de estructura intermediaria: como se demuestra más
adelante (Sec. 5.14), reactivos y
producto son tetraédricos en reactivos SN2, mientras que su estado
de transición contiene carbono pentavalente.)
En la sección 2.18 estudiamos las velocidades de
reacción desde el punto de vista de la teoría de las colisiones. Un enfoque
alternativo de utilidad más general es la
teoría del estado de transición (o
termodinámica) de la velocidad de reacción. Se considera que existe un
equilibrio entre los reactivos y el estado de transición, el cual se trata en
la misma forma que los equilibrios verdaderos de reacciones reversibles (Sec.
23.11). Se reemplazan la energía de activación (Eact) y el factor de
probabilidad por calor de activación (entalpía, DH++)
y entropía de activación (DS++),
respectivamente, los que, combinados, dan la energía libre de activación (DG++).
DG++ = DH++ - TDS++
Cuanto menor (menos positivo) sea el DH++
y cuanto mayor (más positivo) el DS++,
menor será DG++ y más rápida la reacción.
La entropía corresponde, aproximadamente, al grado
de desorden de un sistema: un equilibrio tiende a favorecer el estado en el
cual se imponen menos restricciones a los átomos y a las moléculas. Luego, la
entropía de activación es una medida del desorden relativo de reactivos y
estado de transición; cuanto menos restricciones a la disposición de los átomos
en el estado de transición -en relación con los reactivos -, más rápida será la
reacción. Podemos apreciar que, en
general, el factor de probabilidad y la entropía de activación miden prácticamente lo mismo; por una parte, un
factor de probabilidad bajo significa que en una colisión se requiere una
orientación muy especial de los átomos; por otra parte, una entropía de
activación desfavorable (baja) significa que se imponen restricciones bastante
severas en cuento a las posiciones de los átomos en el estado de transición.
2.24 Reactividad y desarrollo del estado de transición
Para la separación de hidrógeno del metano por
medio de un átomo de halógeno, acabamos
de ver que el estado de transición difiere de los reactivos diferencia que,
desde luego, estamos investigando - en el sentido de que se parece a los
productos. Esto es generalmente cierto para reacciones en las que se forman
radicales libres (o iones carbonio o carbaniones).
Pero, ¿cuánto
se parece este estado de transición en particular a los productos? ¿Hasta dónde han llegado la ruptura y formación de
enlaces? ¿En qué grado se ha hecho plano el
grupo metilo y en qué proporción tiene el electrón no apareado?
Sorprendentemente, podemos incluso contestar a preguntas como éstas, al menos en forma relativa. En un grupo de reacciones similares, el estado de transición se alcanza
tanto más tarde, durante el proceso, cuanto más
alta sea la Eact. De las consideraciones teóricas que
fundamentalmente este postulado, sólo mencionaremos ésta: la diferencia en
distribución electrónica que llamamos
diferencia en estructura corresponde a una diferencia energética; cuanto mayor sea la
diferencia estructural, mayor será la
energética. Si Eact es elevada, el estado de transición difiere considerablemente en
energía de los reactivos y tal vez también en estructura electrónica; si Eact es baja,
habrá poca diferencia entre la energía del estado de transición y la de los
reactivos, como probablemente también se estructura electrónica (véase Fig.
2.12).
En la práctica, este postulado ha resultado muy
útil para la interpretación de
resultados experimentales; veremos que, otros, nos permite explicar la relación
entre reactividad y selectividad (Sec. 3.28).
La separación de hidrógeno por el átomo de cloro,
muy reactivo, tiene una baja Eact. Luego, de acuerdo con el
postulado, se alcanza el estado de
transición antes de que la reacción haya
avanzado mucho y cuando el enlace
carbono-hidrógeno se encuentra sólo
ligeramente estirado; la distribución de átomos y electrones aún es muy
similar a la de los reactivos, y el carbono es todavía prácticamente
tetraédrico. El grupo metilo ha desarrollado poco carácter de radical libre.
En cambio, la separación de hidrógeno por el átomo de bromo, menos
reactivo, tiene una Eact muy elevada. El estado de transición sólo se alcanza una vez
avanzada la reacción hasta estar cerca de completarse y cuando el enlace
carbono-hidrógeno está casi escindido.
La geometría y la distribución electrónica han
comenzado a aproximarse a la de los productos y el carbono bien puede ser casi
trigonal. El grupo metilo ha desarrollado mucho carácter de radical libre.
Así, en
el ataque por un reactivo de gran reactividad, el estado de transición tiende a
parecer al reactivo; en el ataque por una sustancia de poca reactividad, el
estado de transición tiende a ser semejante a los productos.
2.25 Fórmula molecular: su importancia fundamental
En este capítulo nos hemos ocupado de la estructura del metano: el modo de
juntarse los átomos para formar la molécula de metano. Antes, sin embargo, es
necesario conocer de qué átomos se trata
y cuántos de ellos conforman la molécula: es primordial saber que
el metano es CH4. Antes de
poder asignar una fórmula estructural a un compuesto, debemos conocer su
fórmula molecular.
Se ha invertido mucho de este capítulo en el
estudio de la sustitución del cloro por el hidrógeno en el metano, pero
antes fue necesario saber que había
sustitución, que cada paso de la reacción genera un producto que contiene un
hidrógeno menos y un átomo de cloro más
que el reactivo; debíamos saber que el CH4 es convertido,
sucesivamente, en CH3CI, CH2CI2, CHCI3
y CCI4. Antes de poder estudiar las reacciones de un compuesto
orgánico, debemos conocer las fórmulas moleculares de los productos.
Revisemos un poco lo que sabemos acerca de cómo
asignar una fórmula molecular a un compuesto. Debemos realizar:
(a) un
análisis elemental cualitativo, para determinar que tipos de átomos
contiene la molécula;
(b) un
análisis elemental cuantitativo, para determinar el número relativo de los
distintos tipos de átomos presentes en la molécula, es decir, para establecer
su fórmula empírica;
(c) una
determinación del peso molecular, que indica (combinado con la fórmula
empírica) el verdadero número de los distintos átomos, es decir, nos da la
fórmula molecular.
La mayor parte de esto debería serle familiar al
estudiante de cursos anteriores de química. Aplicaremos estos principios al
análisis orgánicos.
2.26 Análisis elemental cualitativo
En una sustancia, la presencia de carbono e
hidrógeno se detecta por combustión:
un calentamiento con óxido de cobre, que convierte al carbono en dióxido de
carbono y al hidrógeno en agua. (Problema: (Cómo podrían identificarse cada uno de estos productos?)
![]()
Un halógeno, el nitrógeno y el azufre, enlazados
covalentemente, deben convertirse en iones inorgánicos, que pueden ser detectados
por métodos ya conocidos. Puede lograrse esta conversión por cualquiera de
estos dos caminos: (a) por medio de la fusión
con sodio, un tratamiento con sodio metálico fundido;
![]()
o (b) por la oxidación
de Schoniger con oxígeno gaseoso.
![]()
(Un método más simple para detectar un halógeno en
algunos compuestos orgánicos se
estudia más adelante en la Sec. 5.26.)
Con estos métodos podríamos demostrar, por ejemplo, que el metano
contiene carbono e hidrógeno, o que el cloruro de metilo contiene carbono,
hidrógeno y cloro.
Pruebas adicionales han demostrado la ausencia de
cualquier otro elemento en estos compuestos, salvo posiblemente oxígeno, para
el que no hay ensayo químico simple; su presencia o ausencia se demuestra por
un análisis cuantitativo.
2.27 Análisis elemental cuantitativo: carbono, hidrógeno
y halógeno
Conocidos los
elementos que conforman un compuesto, debemos determinar las proporciones en
que se encuentran. Para lograr esto, efectuamos prácticamente el mismo análisis
que antes, pero ahora de forma
cuatitativa. Por ejemplo, para encontrar
las cantidades relativas de carbono e hidrógeno en el metano, oxidaríamos completamente una cantidad medida
de metano y pesaríamos el dióxido de carbono y el agua formados.
En una combustión cuantitativa, se hace pasa una
muestra pesada del compuesto orgánico por un tren de combustión: éste
es un tubo, lleno de óxido de cobre y calentado de 600ºC a 800ºC, seguido de
otro tubo que contiene agente desecante (generalmente Dehidrita, perclorato de
magnesio) y de otro cargado con una
base fuerte (generalmente Ascarita,
hidróxido de sodio sobre asbesto). El
agua formada se absorbe en el
desecante, y el dióxido de carbono, en
la base; el aumento de peso en cada uno de estos tubos indica el peso del
producto formado.
Podríamos haber determinado, por ejemplo, que una muestra metano de 9.67 mg de peso produjo 26.53 mg de CO2 y 21.56 mg de H2O. Ahora, solamente la fracción C/CO2=12.01/44.01 del dióxido de carbono es carbono y sólo la fracción 2H/H2O=2.016/18.02 de agua es hidrógeno. En consecuencia,
![]()
y la composición porcentual es
![]()
Puesto que el total de carbono e hidrógeno es
100%, dentro de los límites de error del análisis, no puede haber oxígeno (ni
ningún otro elemento).
Como en el análisis cualitativo, en el
cuantitativo el halógeno unido covalente debe convertirse en ión halogenuro. Se
calienta el compuesto orgánico, (a) en una bomba con peróxido de sodio o (b) en
un tubo sellado con ácido nítrico (método
de Carius). El ión halogenuro así formado se convierte en halogenuro de
planta, el cual se puede pesar.
(Adoptaremos otros métodos analíticos
cuantitativos cuando los necesitemos: análisis de nitrógeno y azufre, Sec.
13.12; equivalente de neutralización, Sec. 23.21; índice de saponificación,
Sec. 24.24.)
2.28 Fórmula empírica
Conociendo la composición porcentual de un
compuesto, podemos calcular la fórmula
empírica: la fórmula más simple que
indica los números relativos de los diferentes tipos de átomos en una molécula.
Por ejemplo, en 100 g (se toman por conveniencia) de metano, hay 74.9 g de
carbono y 24.9 g de hidrógeno, de acuerdo con nuestro análisis cuantitativo.
Dividiendo cada cantidad entre el peso atómico apropiado, se obtiene el número
de moles de cada elemento.
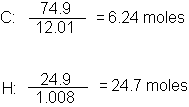
Puesto que un mol de un elemento contiene el mismo
número de átomos que un mol de cualquier otro elemento, sabemos ahora el número
relativo de átomos de carbono e hidrógeno en el metano: C6.24H24.7.
La conversión a los números enteros más pequeños da su fórmula empírica CH4
para el metano.
C: 6.24/6.24 = 1
H: 24.7/6.24 = 3.96, aproximadamente 4
2.29 Peso molecular. Fórmula molecular
Sabemos ahora qué átomos conforman la molécula que
estudiamos y en qué proporciones se encuentran , lo que se resume en la fórmula
empírica.
Esto no es suficiente, sin embargo; basándose
solamente en su fórmula empírica, por ejemplo, el metano podría tener un
carbono y cuatro hidrógenos, o dos carbonos y ocho hidrógenos, o cualquier múltiplo de CH4.
Aún nos resta encontrar la fórmula
molecular: que indica el número
verdadero de cada clase de átomo en una molécula.
Para encontrar la fórmula molecular, debemos
determinar el peso molecular: hoy seguramente se haría por espectrometría de
masas, la que da un valor exacto (Sec. 16.2). El etano, por ejemplo, tiene la
fórmula empírica CH3: se le encuentra un peso molecular de 30, lo
que indica que C2 H6 debe ser la única fórmula molecular
correcta entre todas las posibles.