1.- Estructuras y propiedades
(Ir a página principal http://organica1.org/ )
1.3 El
enlace químico antes de 1926
1.6 Configuración
electrónica. Principio de exclusión de Pauli
1.12 Pares de
electrones no compartidos.
1.14 Energía de
disociación de enlace. Homólisis y heterólisis
1.16
Polaridad de las moléculas
1.17
Estructura y propiedades físicas
1.1 Química
orgánica
La
química orgánica es la química de los compuestos de carbono.
El nombre engañoso <<orgánico>> es una
reliquia de los tiempos en que los compuestos químicos se dividían en dos
clases: inorgánicos y orgánicos, según su procedencia. Los compuestos
inorgánicos eran aquellos que procedían de los minerales, y los orgánicos, los
que se obtenían de fuentes vegetales y animales, o sea, de materiales
producidos por organismos vivos. De hecho, hasta aproximadamente 1850 muchos
químicos creían que los compuestos orgánicos debían tener su origen en
organismos vivos y, en consecuencia, jamás podrían ser sintetizados a partir de
sustancias inorgánicas.
Los compuestos de fuentes orgánicas tenían en común lo
siguiente: todos contenían el elemento carbono. Aun después de haber quedado
establecido que estos compuestos no tenían necesariamente que proceder de
fuentes vivas, ya que podían hacerse en el laboratorio, resultó conveniente
mantener el nombre orgánico para
describir éstos y otros compuestos
similares, persistiendo hasta la fecha esta división entre compuestos
inorgánicos y orgánicos.
Aunque aún hoy muchos compuestos del carbono se aíslan
mejor a partir de fuentes vegetales y animales, la mayoría de ellos se obtienen por síntesis. A veces se
sintetizan a partir de sustancias inorgánicas, como carbonatos y cianuros, pero
más a menudo se parte de otros compuestos orgánicos. Hay dos grandes fuentes de
las que se pueden obtener sustancias orgánicas simples: el petróleo y el carbón.
(Ambas son <<orgánicas>> en el sentido
tradicional, puesto que son producto de la descomposición de plantas y animales.) Estas sustancias simples se emplean como
elementos básicos, a partir de los
cuales se pueden hacer compuestos más complicados.
Reconocemos al petróleo y al carbón como combustibles fósiles, acumulados
durante milenios y no renovables que se
están consumiendo a una velocidad alarmante, en particular el petróleo, para
satisfacer nuestra siempre creciente demanda de energía. Hoy, menos del 10% del
petróleo utilizado se consume en la
fabricación de productos químicos; la mayor parte, sencillamente, se quema para
proporcionar energía. Afortunadamente, existen otras fuentes de energía: la
solar, la geotérmica y la nuclear, pero ¿dónde
habremos de encontrar una reserva sustitutiva de materias orgánicas?
Tarde o temprano, por supuestos, tendremos que volver al lugar de donde
proceden originalmente los combustibles
fósiles -la biomasa- aunque ahora directamente, prescindiendo de los milenios
que intervinieron. La biomasa es renovable y, utilizada adecuadamente, puede
perdurar en este planeta tanto como nosotros mismos. Mientras tanto, se ha
sugerido que el petróleo es demasiado valioso para ser quemado.
¿Qué tienen de
especial los compuestos del carbono que justifique su separación de los otros
ciento y pico elementos de la tabla periódica? Al menos parcialmente, la respuesta parece ser ésta:
hay muchísimos compuestos del carbono, y sus moléculas pueden ser muy grandes y
complejas.
El número de compuestos que contienen carbono es muchas
veces mayor que el número de los que no lo contienen. Estos compuestos
orgánicos se han dividido en familias que, en general, no tienen equivalentes
entre los inorgánicos.
Se conocen moléculas orgánicas que contienen miles de
átomos, cuyo ordenamiento puede ser muy complicado, aun en moléculas
relativamente pequeñas. Uno de los principales problemas en química orgánica es encontrar cómo se ordenan los átomos en las moléculas, o sea, determinar las
estructuras de los compuestos.
Hay muchas maneras en que estas complicadas moléculas pueden romperse o reordenarse para generar
moléculas nuevas; hay muchas formas de agregar átomos a estas moléculas o de
sustituir átomos nuevos por antiguos. Una parte importante de la química
orgánica se dedica a encontrar estas reacciones, cómo suceden y cómo pueden
emplearse para sintetizar las sustancias que queremos.
¿Que tiene de especial el carbono para formar tantos
compuestos? La respuesta a esta pregunta se le ocurrió a August Kekulé en 1854 durante un viaje en ómnibus en
Londres.
<<Era una
noche de verano. Regresaba en el último
ómnibus absorto como siempre, por las calles
desiertas de la ciudad, que a otras horas están llenas de vida. De
pronto los vi, los átomos danzaban ante mis ojos... Vi cómo, frecuentemente,
dos pequeños átomos se unían formando un par; vi cómo uno más grande aceptaba
dos más pequeños; cómo uno aún mayor sujetaba
a tres e incluso a cuatro de loa más pequeños, mientras el conjunto
continuaba arremolinándose en una danza vertiginosa. Vi cómo los más grandes formaban
una cadena... Pasé parte de la noche vertiendo al papel algunos esbozos de
estas formas soñadas.>> (
August Kekulé, 1890.)
Los átomos de carbono pueden unirse entre sí hasta grados
imposibles para los átomos de cualquier
otro elemento. Pueden formar cadenas de miles de átomos o anillos de
todos los tamaños; estas cadenas y anillos pueden tener ramificaciones y uniones cruzadas. A los carbonos de estas cadenas y anillos se unen otros
átomos ; principalmente de hidrógeno, pero también de flúor, cloro, bromo, yodo, oxígeno,
nitrógeno, azufre, fósforo y muchos otros.
[Véase,
a modo de ejemplos, la celulosa (Sec. 39.11), la clorofila (Sec. 35.1) y la
oxitocina (sec. 40.8).]
Cada ordenamiento atómico diferente corresponde a un
compuesto distinto, y cada compuesto tiene su conjunto de características
químicas y físicas. No es sorprendente que hoy se conozcan cerca de diez
millones de compuestos del carbono y que este número aumente en medio millón cada año. No es de
sorprender que el estudio de su química sea un campo especializado.
La química orgánica es un campo inmensamente importante
para la tecnología: es la química de los colorantes y las drogas, del papel y
las tintas, de las pinturas y los plásticos, de la gasolina y lo neumáticos; es
la química de nuestros alimentos y de
nuestro vestuario.
La química orgánica es fundamental para la biología y la medicina. Los organismos vivos están
constituidos principalmente por sustancias orgánicas, además de agua; las
moléculas de la <<biología
molecular>> son
orgánicas. A nivel molecular, la biología es química orgánica.
Parte 2
1.2 La teoría estructural
<<La
química orgánica actual está a punto de enloquecerme. Se me figura como un
bosque tropical primigenio lleno de las cosas más notables, una selva infinita y
terrible en la que uno no se atreve a penetrar porque parece que no hay salida.
>> (Friedrich Wohler, 1835.)
¿Cómo podemos siquiera comenzar el estudio de una materia
tan enormemente compleja? ¿Es hoy la química orgánica como la veía Wohler hace siglo y medio? La selva aún está ahí- en
gran parte inexplorada- y en ella hay
cosas mucho más notables que las
que Wohler puedo haber soñado. Sin embargo, mientras no vayamos demasiado
lejos, ni demasiado aprisa, podremos penetrar en ella sin el temor a perdernos,
pues tenemos un mapa: la teoría estructural.
La teoría estructuras es la base sobre la cual se han acumulado millones de hechos
acerca de cientos de miles de compuestos individuales, ordenándolos en forma
sistemática. Es la base sobre la cual estos hechos pueden explicarse y
comprenderse mejor.
La teoría estructural es el marco de ideas acerca de cómo
se unen los átomos para formar moléculas. Tiene que ver con el orden en que se
juntan los átomos y con los electrones que los mantienen unidos. Tiene que ver
con las formas y tamaños de las moléculas que generan estros átomos y con el modo de distribución de los
electrones a su alrededor.
A menudo se presenta una molécula por un dibujo o un
modelo; a veces por varios dibujos o varios modelos. Los núcleos atómicos se presentan por letras o esferas
de plástico, y los electrones que los unen, por líneas, punto o varillas de
plásticos. Estos modelos y dibujos aproximados son útiles para nosotros sólo si
entendemos lo qué representan. Interpretados en función de la teoría
estructural, nos revelan bastante acerca del compuesto cuyas moléculas
representan; cómo proceder para hacerlo, qué propiedades físicas se pueden
esperar de él- punto de fusión, punto
de ebullición, densidad, tipo de disolventes en que se disolverá el compuesto,
si será coloreado o no, qué tipo de comportamiento químico esperar-, la clase de reactivos con los que reaccionará y el tipo de productos que formará, y si
reaccionará rápida y lentamente. Se
podría saber todo esto acerca de un compuesto desconocido para nosotros
simplemente partiendo de su fórmula
estructural y de los que entendemos
que ésta significa.
Parte 3
1.3 El enlace
químico antes de 1926
Toda consideración de la estructura de las moléculas debe
comenzar con un estudio de los enlaces
químicos, las fuerzas que mantienen unidos a los átomos en una molécula.
Estudiaremos los enlaces químicos en función de la teoría desarrollada antes de
1926, y luego en función de la teoría actual. La introducción de la mecánica
cuántica en 1926 provocó un gran cambio
en las ideas sobre la formación de las moléculas. Por conveniencia, aún suelen emplearse las representaciones pictóricas y el
lenguaje iniciales, más simples. Dándoles una interpretación moderna.
En 1916 se describieron dos clases de enlace químico: el enlace iónico, por Walter Kossel (Alemania), y el enlace covalente, por G. N. Lewis ( de la Universidad de
California).
Tanto Kossel como Lewis basaron sus ideas en el siguiente
concepto del átomo.
Un núcleo cargado
positivamente está rodeado de
electrones ordenados en capas o niveles energéticos concéntricos. Hay un máximo
de electrones que se pueden acomodar en
cada capa: dos en la primera, ocho en
la segunda, ocho o dieciocho en la tercera, y así sucesivamente. La estabilidad
máxima se alcanza cuando se completa la
capa externa, como en los gases nobles. Tanto los enlaces iónicos como los
covalentes surgen de la tendencia de los átomos a alcanzar
esta configuración electrónica estable.
El enlace iónico
resulta de la transferencia de
electrones, como, por ejemplo, en la formación del fluoruro de litio. Un
átomo de litio tiene dos electrones en
su capa interna y uno en su capa externa o de valencia; la
pérdida de un electrón dejaría al litio con una capa externa completa de dos
electrones. Un átomo de flúor tiene dos electrones en su capa interna y siete
en su capa de valencia; la ganancia de un electrón daría el flúor una capa
externa completa con ocho electrones.
El fluoruro de litio se forma por la transferencia de un electrón del litio al
flúor; el litio tiene ahora una carga positiva, y el flúor, una negativa. La
atracción electrostática entre iones de carga opuesta se denomina enlace
iónico, el cual es típico en las sales
formadas por combinación de elementos metálicos (elementos
electropositivos) del extremo izquierdo
de la tabla periódica con los elementos no metálicos ( elementos electronegativos) del extremo derecho.
El enlace
covalente resulta de compartir
electrones, como, por ejemplo, en la formación de la molécula de hidrógeno.
Cada átomo de hidrógeno tiene un solo electrón; al compartir un par de electrones, ambos hidrógenos
pueden completar sus capas de
dos. Dos átomos de flúor, cada uno con siete electrones. De forma similar,
podemos visualizar la formación de HF,
H2O, NH3, CH4 y CF4. También
aquí la fuerza de unión es la atracción electrostática, esta vez entre cada
electrón y ambos núcleos.
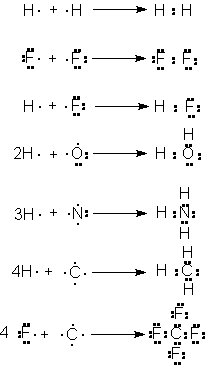
El
enlace covalente es típico de los
compuestos del carbono; es el enlace de mayor importancia en el estudio de la
química orgánica.
Parte 4
1.4 Mecánica
cuántica
En 1926 salió a la luz la teoría conocida como mecánica cuántica, cuyo desarrollo, en
la forma más útil para los químicos, se debe a Erwin Schrodinger (de la Universidad de Zurich), que desarrolló
expresiones matemáticas para describir el movimiento de un electrón en función de su energía. Estas expresiones matemáticas
se conocen como ecuaciones de onda, puesto que se basan en el
concepto de que el electrón no sólo presenta propiedades de partículas, sino
también de ondas.
Una ecuación de onda tiene diversas soluciones,
llamadas funciones de onda, y cada una corresponde a un nivel de energía
diferente para el electrón.
Salvo para los sistemas más simples, las matemáticas correspondientes a la
obtención de soluciones consumen tanto tiempo esto lo cambiarán algún día los computadores superveloces que sólo es posible obtener soluciones aproximadas. Aun así, la
mecánica cuántica da respuestas que
concuerdan tan bien con los hechos que
es aceptada hoy día como la herramienta más útil para la comprensión de las
estructuras atómica y molecular.
<<La
mecánica ondulatoria nos ha indicado lo que está sucediendo, y al nivel más profundo posible... ha tomado los conceptos del químico experimental la percepción imaginativa que poseían
quienes vivieron en sus laboratorios y que permitieron que sus mentes recrearan creativamente los hechos que habían
descubierto y ha demostrado cómo todos
encajaban; cómo, si se quiere, todos ellos presentan una sola lógica, y cómo pueden
develarse esta relación escondida entre ellos.>> (C. A. Coulson. Londres,
1951.)
Parte 5
1.5 Orbitales atómicos
Una ecuación de onda no puede indicarnos exactamente el
lugar en que se encuentra un electrón en un instante particular ni lo rápido
que se está moviendo; no nos permite dibujar
una órbita precisa en torno al núcleo. En cambio, nos revela la probabilidad de encontrar el electrón en
cualquier lugar particular.
La
región en el espacio en la que es probable
que se encuentre un electrón se
denomina orbital. Hay diferentes tipos de orbitales, con tamaño y formas
diferentes, y que están dispuestos en
torno al núcleo de maneras específicas. El tipo particular de orbital que ocupa un electrón depende de
su energía. Nos interesan especialmente las formas de estos orbitales y sus posiciones recíprocas, puesto que
determinan o, más exactamente, puede considerarse que determinan las disposición espacial de los átomos de una
molécula e incluso ayudan a determinar
su comportamiento químico.
Es útil visualizar un electrón como si se difundiera para formar una nube. Esta nube se puede
imaginar como una especie de fotografía
borrosa del electrón en rápido movimiento. La forma de la nube es la forma del orbital. La nube no es
uniforme, sino que es más densa en
aquellas regiones en las cuales la
probabilidad de hallar el electrón es máxima, o sea, en aquellas regiones donde la carga negativa promedio, o densidad electrónica, es máxima.
Veamos cuáles son las formas de algunos orbitales
atómicos. El orbital correspondiente al nivel energético más bajo se denomina 1s,
y es una esfera cuyo centro coincide
con el núcleo del átomo, como se representa en la figura 1.1. Un orbital
no tiene un límite definido, puesto que
hay una probabilidad, aunque muy pequeña, de encontrar el electrón
esencialmente separado del átomo, e incluso sobre otro átomo. Sin embargo, la
probabilidad decrece muy rápidamente más allá de cierta distancia del núcleo,
de modo que la distribución de carga está bastante bien representada por la
nube electrónica de la figura 1.1a. Para simplificar, podemos incluso
representar un orbital como en la figura 1.1b, en la que la línea continua
encierra la región donde el electrón permanece durante la mayor parte del
tiempo (por ejemplo, el 95%).
Fig. 1.1 Orbitales
atómicos: orbital s. El núcleo está en
el centro.
En el nivel energético siguiente se encuentra el orbital
2s, que también es una esfera con su centro en el núcleo atómico, y es naturalmente mayor que el 1s: la mayor
energía (menor estabilidad) se debe a la mayor distancia promedio entre el
electrón y el núcleo, con la consiguiente disminución de la atracción
electrostática. (Considérese el trabajo que debe realizarse la energía a
introducir en el sistema para alejar un electrón del núcleo, que tiene carga
opuesta.)
A continuación
hay tres orbitales de igual energía, llamados orbitales 2p, ilustrados en la
figura 1.2. Cada orbital 2p tiene forma de huso y consta de dos lóbulos entre los cuales está el núcleo atómico.
El eje de cada orbital 2p es perpendicular
a los ejes de los otros dos. Se diferencian por los símbolos 2px,
2py y 2pz, en los que x, y y z son los ejes
correspondientes.
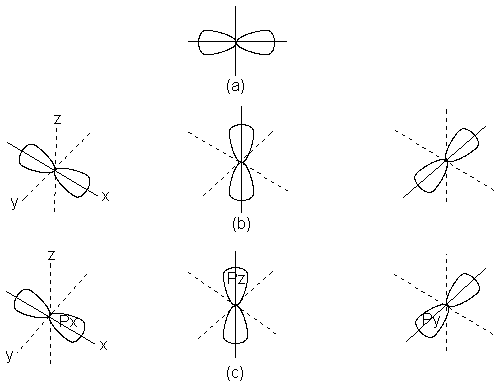
Fig. 1.2 Orbitales atómicos: orbitales p. Ejes
mutuamente perpendiculares.
(a)
Sección transversal mostrando los dos
lóbulos de un orbital individual.
(b)
Forma aproximada de pares de elipsoides distorsionados.
(c)
Representación como pares de esferas que no llegan a tocarse.
Parte 6
1.6 Configuración electrónica. Principio de exclusión de Pauli
Hay una serie de <<reglas>> que determinan el modo
de distribución de los electrones de un átomo, es decir, que determinan la
configuración electrónica de un átomo.
La más fundamental de estas reglas es el principio de exclusión de Pauli: un orbital atómico determinado puede ser
ocupado por sólo dos electrones, que para ello deben tener espines
opuestos. Estos electrones de espines opuestos se consideran apareados. Electrones de igual espín tienden a
separarse lo máximo posible. Esta tendencia es el más importante de los
factores que determinan las formas y propiedades de las moléculas.
El principio de exclusión, desarrollado en 1925 por
Wolfgang Pauli hijo, del Instituto de Física Teórica de Hamburgo
(Alemania), se considera la piedra angular de la química.
Los diez primeros elementos de la tabla periódica tienen las configuraciones electrónicas
indicadas en la tabla 1.1. Podemos apreciar que un orbital no se ocupa hasta
que los orbitales de energía más baja están llenos ( o sea, 2s después de 1s,
2p después de 2s).
Observamos que un
orbital no es ocupado por un par de electrones hasta que otros orbitales de igual energía no sean ocupados
por un electrón ( los orbitales 2p). Los electrones 1s completan la primera
capa de dos, y los electrones 2s y 2p completan la segunda capa de ocho. Para
elementos más allá de los diez primeros hay una tercera capa que contiene un
orbital 3s, orbitales 3p, y así sucesivamente.
Parte 7
1.7 Orbitales moleculares
En las moléculas, al igual que en los átomos aislados, y
de acuerdo con las mismas <<reglas>>, los electrones ocupan
orbitales. Estos orbitales moleculares
se consideran centrados en torno a muchos núcleos, cubriendo quizá la molécula
entera; la distribución de núcleos y electrones es simplemente la que da como
resultado la molécula más estable.
Para facilitar las complicadísimas operaciones
matemáticas, por lo general se emplean dos supuestos simplificadores: (a) que cada par de electrones está localizado esencialmente cerca de dos núcleos solamente
y (b) que las formas de estos
orbitales moleculares localizados, y su
disposición con respecto a los demás, están
relacionadas de modo sencillo con las formas y disposiciones de los orbitales atómicos de los átomos que
componen la molécula.
La idea de los orbitales moleculares localizados o lo que
podríamos llamar orbitales de enlace sin duda es buena, puesto que,
matemáticamente, este método de aproximación es válido para la mayoría de las moléculas (pero no para todas). Además, esta idea se
acerca bastante al concepto clásico de los químicos, según el cual un enlace es
una fuerza que actúa entre dos átomos y
es prácticamente independiente del resto de la molécula; no es accidental que este concepto haya funcionado bien
durante cien años. Es significativo que las moléculas excepcionales, para las cuales las fórmulas clásicas no
funcionan, son justamente las mismas
para las que tampoco sirve el enfoque orbital molecular localizado.
(Veremos que aun estos casos se pueden manejar por medio de una adaptación
bastante sencilla de fórmulas clásicas, una adaptación que también se asemeja a
un método de aproximación matemática.)
El segundo supuesto,
el de una relación entre orbitales atómicos y moleculares, es evidente,
como se apreciará en la siguiente sección. Ha demostrado ser tan útil que, en
ciertos casos, se han inventado
orbitales determinados sólo para poder mantener dicho supuesto.
Parte 8
1.8 El enlace covalente
Consideramos ahora la formación de una molécula. Por
conveniencia, imaginaremos que esto sucede por aproximación de átomos individuales, aunque la mayoría de las moléculas no se forman así. Construimos modelos físicos
de moléculas con esferas de madera o plástico que representan los diversos
átomos; la ubicación de hoyos o broches nos indica cómo unirlos. Del mismo
modo, haremos modelos mentales de
moléculas con átomos imaginarios; la ubicación de los orbitales atómicos
algunos de ellos imaginarios nos indicará cómo unir los átomos.
Para que se forme un enlace covalente, deben ubicarse dos
átomos de manera tal que el orbital de
uno de ellos solape al orbital del
otro; cada orbital debe contener solamente un electrón. Cuando sucede esto,
ambos orbitales atómicos se combinan para formar un solo orbital
de enlace ocupado por ambos electrones, que deben tener espines opuestos,
es decir, deben estar aparecidos. Cada electrón dispone del orbital de enlace
entero, por lo que puede considerarse
como <<perteneciente>> a ambos núcleos
atómicos.
Esta disposición
de electrones y núcleos contiene menos energía es decir, es más estable que
la disposición en los átomos aislados; como resultado, la
formación de un enlace va acompañada de liberación de energía. La cantidad de energía (por mol) desprendida durante la
formación del enlace (o la
cantidad necesaria para romperlo) se
denomina energía de disociación del enlace. Para un par dado de átomos, cuanto mayor sea el solapamiento de orbitales atómicos, más fuerte
será el enlace.
¿Qué es lo que da al enlace covalente su fuerza? Es el aumento de atracción electrostática.
En los átomos aislados, cada electrón es atraído por, y atrae a, un núcleo
positivo; en la molécula, cada electrón es atraído por dos núcleos positivos.
El concepto de <<solapamiento>> es el que proporciona el puente mental entre orbitales atómicos y de enlace.
El solapamiento de orbitales atómicos significa que el orbital de enlace ocupa
gran parte de la región espacial previamente cubierta por ambos orbitales atómicos. En consecuencia, un electrón de un átomo
puede permanecer en gran medida en su ubicación original, favorable con respecto a <<su>> núcleo, y ocupar, al
mismo tiempo, una posición favorable similar con respecto al segundo núcleo;
por supuesto, esto mismo vale para el otro electrón.
El principio de solapamiento
máximo, formulado por primera vez por Linus Pauling en 1931 (Instituto
Tecnológico de California), ha sido clasificado en importancia sólo
ligeramente por debajo del principio de exclusión para la comprensión de
la estructura molecular.
Como primer ejemplo, consideremos la formación de la
molécula de hidrógeno, H2, a partir
de dos átomos. Cada átomo de hidrógeno tiene un electrón, el cual ocupa
el orbital 1s. Como hemos visto, éste es una esfera cuyo centro es el núcleo
atómico. Para que se forme un enlace, ambos núcleos deben acercarse lo
suficiente para que se produzca el
solapamiento de los orbitales atómicos (Fig. 1.3). Para el hidrógeno, el
sistema más estable resulta cuando la distancia entre los núcleos es de 0.74 A,
denominada longitud de enlace. A
esta distancia, el efecto estabilizador del solapamiento es exactamente
compensado por la repulsión entre núcleos de igual carga. La molécula de
hidrógeno resultante contiene 104 kcal/mol menos de energía que los átomos a
partir de los cuales fue construida. Se dice que el enlace hidrógeno-hidrógeno tiene una longitud de 0.74 A y una
fuerza de 104 kcal.
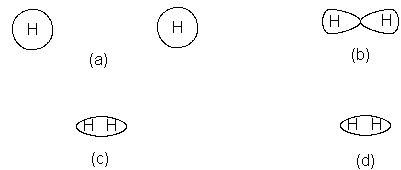
Fig.
1.3 Formación de enlace: molécula de H2. (a) Orbitales s separados.
(b) Solapamiento de orbitales s. (c) y (d) El
orbital de enlace o.
Este orbital de enlace tiene aproximadamente la forma que
se espera obtener de la fusión de dos orbitales s. Tal como indica la figura
1.3, tiene aspecto de salchicha, cuyo
eje mayor coincide con la línea que une los núcleos; en torno a este eje, es
cilíndricamente simétrico, o sea, un corte de
esta salchicha es circular. Los orbitales de enlace que tienen este
aspecto, se denominan orbitales o (orbitales
sigma) y los enlaces correspondientes son los enlaces o. Podemos imaginar
la molécula de hidrógeno como formada por dos núcleos sumergidos en una sola nube electrónica con forma de salchicha.
La densidad máxima de la nube está en
la región entre ambos núcleos, donde la carga negativa es atraída más intensamente por las dos cargas positivas.
El tamaño de la molécula de hidrógeno determinado, por
ejemplo, por el volumen interior de la superficie de probabilidad de 95% es
considerablemente menor que el de un
átomo de hidrógeno individual. Aunque parezca extraño, de hecho es de esperar
esta contracción de la nube electrónica: la intensa atracción que ejercen dos núcleos sobre los electrones
confiere mayor estabilidad a la molécula que la de átomos de hidrógeno
aislados; esto significa que los electrones están sujetos más firmemente,
están más próximos, que en los
átomos.
Supongamos luego la formación de la molécula de flúor, F2,
a partir de dos átomos. Según vemos en la tabla de configuraciones electrónicas
(Tabla 1.1), un átomo de flúor tiene dos elementos en el orbital 1s y dos en
cada uno de dos orbitales 2p; en el tercer orbital 2p hay un solo electrón no
apareado y disponible para formar un enlace. El solapamiento de este
orbital p con uno similar de otro
átomo de flúor permite que los electrones se aparecen y que se forme el
enlace (Fig. 1.4). La carga electrónica
se concentra entre ambos núcleos, de
modo que el lóbulo posterior de cada
uno de los orbitales solapados se
contrae hasta alcanzar un tamaño relativamente pequeño. Aunque formado por
el solapamiento de orbitales a atómicos de diferente tipo, el enlace flúor-flúor tiene la
misma forma general que el enlace hidrógeno-hidrógeno, por ser cilíndricamente
simétrico en torno a la línea de unión
de los núcleos; también se denomina enlace o. El enlace flúor-flúor
tiene una longitud de 1.42 A y una fuerza de unas 38 kcal.
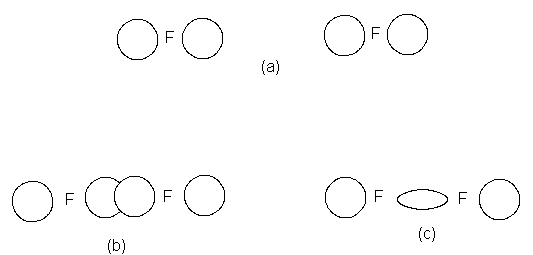
Fig. 1.4 Formación de enlace: molécula de F2.
(a) Orbitales p separados.
(b)
Solapamiento de orbitales p. (c) El orbital de enlace o.
Como indica el ejemplo, un enlace covalente resulta del
solapamiento de dos orbitales atómicos
para formar un orbital de enlace ocupado por un par de electrones. Cada tipo de enlace covalente una longitud y
una fuerza características.
Parte 9
1.9 Orbitales híbridos: sp
A continuación, consideremos el cloruro de berilio, BeCI2.
El berilio (Tabla 1.1) carece de electrones no apareados.
¿Como podemos explicar su combinación con dos átomos de cloro? La formación de
enlaces es un proceso que libera
energía (estabilizante) y tiende a formar enlaces el máximo posible
aunque esto conduzca a orbitales que tengan poca relación con los orbitales
atómicos considerados hasta ahora. Si queremos aplicar aquí nuestro método
mental de construcción de moléculas, habrá que modificarlo. Debemos inventar un
tipo imaginario de átomo de berilio, uno que esté a punto de enlazarse con dos átomos de cloro.
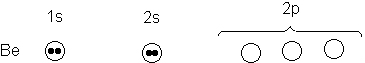
Para llegar a este átomo divalente de berilio, efectuamos
un pequeño cálculo electrónico. En primer lugar, <<promovemos>> uno de los electrones 2s a un orbital p
vacío.
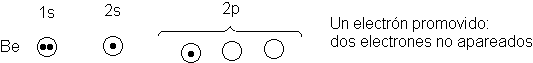
Esto proporciona dos electrones no apareados, necesarios para
enlazar con dos átomos de cloro. Sería
de esperar ahora que el berilio formase un enlace de un tipo empleando el orbital
p y uno de otro tipo con el orbital s. Nuevamente, esto no corresponde a
los hechos: se sabe que los dos enlaces
del cloruro de berilio son
equivalentes.
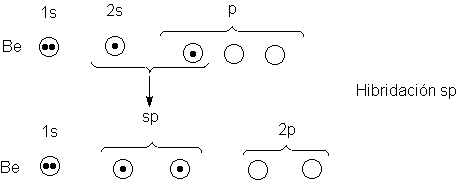
Entonces hibridizemos los orbitales. Tomamos matemáticamente varias posibles combinaciones de un
orbital s y otro p, y se hallan los
orbitales mixtos (híbridos) con el
grado máximo de carácter direccional
(Fig. 1.5). Cuando más se
concentra un orbital atómico
en la dirección del enlace, mayor será
el solapamiento y más
fuerte el enlace que puede formar. De estos cálculos se obtienen tres resultados muy significativos: (a) el <<mejor>> orbital híbrido resulta
mucho más direccional que el orbital s
o el p; (b) los dos orbitales mejores son
exactamente equivalentes, y (c)
estos orbitales apuntan en direcciones opuestas, la disposición que les permite alejarse al máximo entre sí (recuérdese
el principio de exclusión de Pauli). El ángulo entre los orbitales es entonces
de 180º.
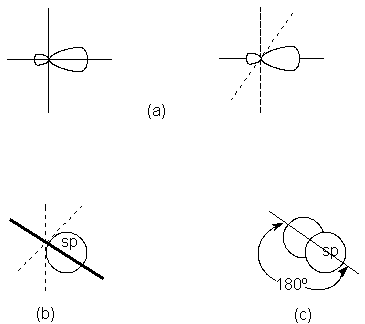
Estos orbitales híbridos específicos se conocen como
orbitales sp, puesto que se consideran como el resultado de la mezcla de un orbital s y uno p, y tienen la forma indicada en la figura 1.5a; por
conveniencia, depreciaremos el pequeño lóbulo posterior y representaremos el
delantero como una esfera.
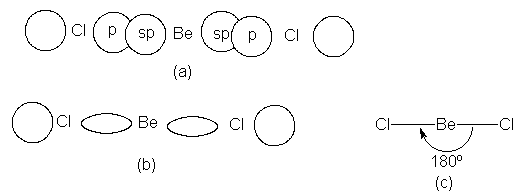
Construyamos el cloruro de berilio usando este berilio
sp-hibridizado. Surge aquí un concepto extremadamente importante: el ángulo de enlace. Para lograr el
solapamiento máximo entre los orbitales sp del berilio y los p de los cloruros,
los dos núcleos de cloro deben encontrarse sobre los ejes de los orbitales
sp, es decir, deben estar localizados
en lados exactamente opuestos del átomo de berilio (Fig. 1.6). Por
tanto, el ángulo entre los enlaces berilio-cloro debe ser de 180º.
Experimentalmente, se ha demostrado que, según lo
calculado, el cloruro de berilio es una
molécula lineal, con los tres átomos ubicados sobre una sola línea recta.
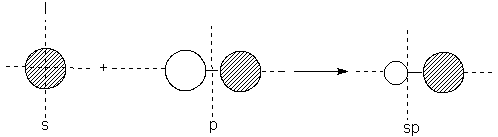
No hay nada de mágico en el aumento del carácter direccional
que acompaña a la hibridación; los dos lóbulos del orbital p son de fase opuesta (Sec. 33.2); la combinación con un orbital s significa adición a un lado del núcleo, pero sustracción en el otro.
Si se tiene curiosidad con respecto a las fases y su
efecto sobre los enlaces, léanse las secciones 33.1 a 33.4, que permitirán
entender este punto.
Parte 10
1.10 Orbitales híbridos: sp2
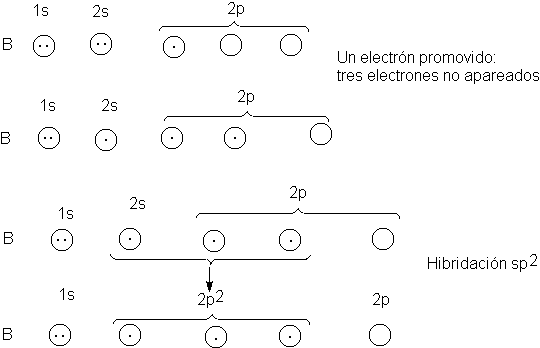
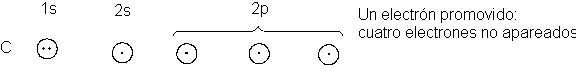
Veamos ahora el trifluoruro de boro, BF3. El
boro (Tabla 1.1) tiene sólo un electrón no apareado, que ocupa un orbital 2p.
Para tres enlaces necesitamos tres electrones no apareados, por lo que
promovemos uno de los electrones 2s a un orbital 2p:
Si ahora queremos <<construir>> la molécula más estable
posible, debemos <<hacer>> los enlaces más fuertes
posibles, para lo que hay que proporcionar
los orbitales atómicos más intensamente direccionales que se pueda.
Nuevamente, la hibridación nos proporciona tales orbitales: tres de ellos híbridos
y exactamente equivalente entre sí. Cada uno tiene la forma indicada en la
figura 1.7 y, como antes, despreciaremos el pequeño lóbulo posterior y
representaremos el delantero como una esfera.
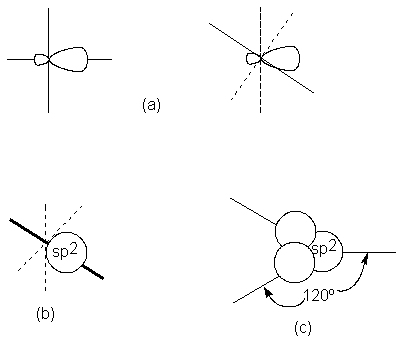
Estos orbitales híbridos se llaman sp2, debido
a que se consideran generados por la mezcla de un orbital s y dos orbitales p.
Se encuentran en un plano que incluye el núcleo atómico y dirigidos hacia los
vértices de un triángulo equilátero, de modo que el ángulo entre dos orbitales
cualesquiera es de 120º. Nuevamente, observamos la geometría que permite la
separación máxima posible de los orbitales: en este caso, es una disposición trigonal ( de tres vértices).
Cuando ordenamos los átomos para lograr el solapamiento
máximo de cada uno de los orbitales sp2 del boro con un orbital p
del flúor, obtenemos la estructura ilustrada en la figura 1.8: una molécula plana, con el átomo de boro en
el centro de un triángulo y los tres átomos de flúor en los vértices; cada
ángulo de enlace es de 120º.
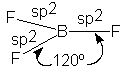
Por experimentación se ha demostrado que el fluoruro de
boro tiene esta estructura plana y simétrica calculada por mecánica cuántica.
Parte 11
1.11 Orbitales
híbridos: sp3
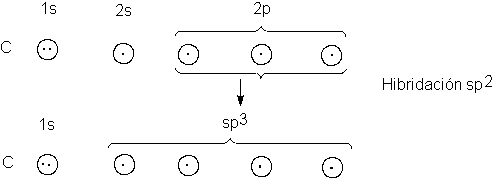
Consideremos ahora una de las moléculas orgánicas más
simples, el metano, CH4.
El carbono (Tabla
1.1) tiene un electrón no apareado en cada uno de los dos orbitales p, por lo
que sería de esperar que formara el compuesto CH2. (Lo forma, pero el CH2 es una
molécula altamente reactiva cuyas propiedades se centran en torno a la
necesidad de procurarle al carbono dos
enlaces adicionales.) Observamos nuevamente la tendencia a formar el máximo posible
de enlaces; en este caso, la combinación con cuatro átomos de hidrógeno.

Para disponer de cuatro electrones no apareados,
promovemos uno de los electrones 2s a un orbital p vacío:
Una vez más, los orbitales más intensamente direccionales
son híbridos: esta vez son orbitales sp3, que resultan de la mezcla
de un orbital s y tres p. Cada uno
tiene la forma ilustrada en la figura 1.9; tal como hemos hecho con los
orbitales sp y sp2, despreciaremos al pequeño lóbulo posterior y
representaremos el delantero por medio de una esfera.
¿Qué disposición espacial tienen los orbitales sp3?
Para nosotros, la respuesta no es una sorpresa: aquella que les permite
separarse al máximo. Se dirigen hacia los vértices de un tetraedro regular. El
ángulo entre dos orbitales cualesquiera es el tetraédrico de 109.5º
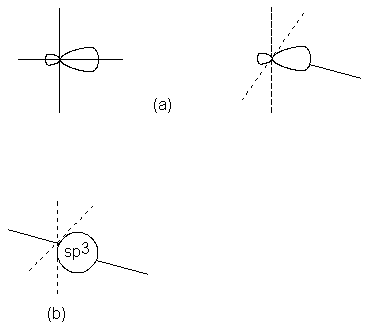
(Fig. 1.9). AI igual que genera dos enlaces lineales o
tres trigonales, la repulsión mutua entre orbitales también genera cuatro
enlaces tetraédricos.
El solapamiento de cada uno de los orbitales sp3
del carbono con un orbital s del hidrógeno genera metano, con el carbono en el
centro de un tetraedro regular y los cuatro hidrógenos en los vértices (Fig.
1.10).
Se ha encontrado experimentalmente que el etano tiene la
estructura altamente simétrica que hemos armado. Cada enlace carbono-hidrógeno
tiene exactamente la misma longitud, 1.10 A; el ángulo entre cualquier par de
enlaces es el tetraédrico de 109.5º. Se necesitan 104 kcal/mol para romper uno
de los enlaces del metano.
Así pues, en estas tres últimas secciones hemos visto que
con los enlaces covalentes no sólo están asociadas longitudes y energías de
disociación de enlaces características, sino también ángulos de enlace
característicos; estos enlaces pueden relacionarse sin dificultad con la
disposición de los orbitales atómicos incluyendo los híbridos que intervienen
en la formación de los enlaces y se
derivan por último del principio de exclusión de Pauli y de la tendencia de los electrones no apareados a
separarse al máximo.
A diferencia del enlace iónico, igualmente fuerte en
todas las direcciones, el enlace
covalente es dirigido. Podemos comenzar a ver por qué la química del enlace
covalente se ocupa tanto de la forma y
el tamaño moleculares.
Dado que los compuestos del carbono están unidos
principalmente por enlaces covalentes, la química orgánica también está muy
interesada en la forma y el tamaño moleculares; para ayudarnos en su estudio, utilizaremos con frecuencia modelos
moleculares. En la figura 1.11 se observa
el metano representado por tres tipos diferentes de modelos: esferas y
palillos, armazón y semiesferas. Estas
últimas están hechas a escala y reflejan
con exactitud no solamente los ángulos
de enlace, sino también sus longitudes relativas y el tamaño de los
átomos.
Parte 12
1.12 Pares de electrones no compartidos.
Dos compuestos conocidos, el amoniaco (NH3) y
el agua (H2O), ilustran cómo pares
de electrones no compartidos pueden afectar a la
estructura molecular.
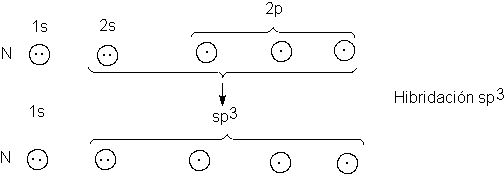
El nitrógeno del amoniaco se asemeja al carbono del metano: tiene hibridación sp3,
pero tiene solamente tres electrones no apareados (Tabla 1.1), que ocupan tres
de los orbitales sp3. El solapamiento de cada uno de esos orbitales
con el orbital s de un átomo de hidrógeno genera amoniaco (Fig. 1.12). El
cuarto orbital sp3 del nitrógeno contiene un par de electrones.
Si ha de haber solapamiento máximo, y por tanto fuerza
máxima de enlace, los tres núcleos de hidrógeno deben localizarse en tres
vértices de un tetraedro, mientras que el cuarto deberá ser ocupado por un par de electrones no compartido. Si se
consideran sólo núcleos atómicos, la
molécula de amoniaco debería tener forma piramidal, con el nitrógeno en el ápice y los hidrógenos en los
vértices de una base triangular. Cada ángulo de enlace debería ser el
tetraédrico de 109.5º.
Se ha encontrado experimentalmente que el amoniaco tiene
la forma piramidal calculada por mecánica cuántica. Los ángulos de enlace son
de 107º, ligeramente menores que el
valor predicho, por lo que se ha sugerido que el par de electrones no
compartido ocupa más espacio que cualquiera de los átomos de hidrógeno,
tendiendo así a comprimir ligeramente los ángulos de enlace. La longitud del enlace nitrógeno-hidrógeno es de 1.01
A; se necesitan 103 kcal/mol para romper uno de los enlaces
del amoniaco.
El orbital sp3 ocupado por el par de
electrones no compartido es una región de alta densidad electrónica. Esta
región es una fuente de electrones para átomos y moléculas que los buscan, lo
que confiere al amoniaco sus propiedades básicas ( Sec. 1.22).
Pueden concebirse dos configuraciones adicionales para el
amoniaco, pero ninguna satisface los hechos.
(a) Como el
nitrógeno está unido a otros tres átomos, podríamos haberlo concebido
utilizando orbitales sp2,
como hace el boro en el trifluoruro de boro. Pero el amoniaco no es una
molécula plana, por lo que debemos
rechazar esta posibilidad. El par de electrones no compartido del nitrógeno es
el responsable de la diferencia entre
el NH3 y el BF3; estos electrones necesitan alejarse de
los que están en los enlaces carbono-hidrógeno,
y la forma tetraédrica lo hace posible.
(b) Podríamos
haber imaginado al nitrógeno empleando
simplemente los orbitales p para el
solapamiento, puesto que proporcionarían los tres electrones no
apareados necesarios; pero esto generaría ángulo de enlace de 90º - recuérdese
que los orbitales p son perpendiculares entre sí -, en contraste con los ángulos observados de
107º. Más importante aún es que el par no compartido se encontraría sumergido
en un orbital s, y por los momentos
dipolares (Sec. 1.16) se envidia que no
es así. Es evidente que la
estabilidad ganada por el empleo de los orbitales sp3 fuertemente
direccionales en la formación de enlaces compensa sobradamente la promoción de
un par no compartido de un orbital s a
otro sp3 más energético.
Un hecho adicional acerca del amoniaco, es que la
espectroscopia revela que la molécula
sufre inversión, es decir, se
vuelve de dentro afuera (Fig. 1.13).
Entre una disposición piramidal y la otra equivalente hay una barrera energética
de sólo 6 kcal/mol, energía que es proporcionada por colisiones moleculares;
aun a temperatura ambiente, la fracción
de colisiones suficientemente violentas para realizar la tarea es tan grande que la conversión entre disposiciones piramidales sucede con gran velocidad.
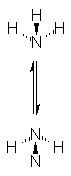
Fig.
1.13 Inversión del amoniaco.
Compárese el amoniaco con el metano, el cual no sufre
inversión. El par no compartido desempeña el papel de un enlace
carbono-hidrógeno en la determinación de la forma más estable, la tetraédrica,
de la molécula. Pero, a diferencia de un enlace carbono-hidrógeno, el par no
compartido no puede mantener una disposición tetraédrica particular: una vez apunta en una dirección, y al instante siguiente, en la opuesta.

Finalmente, consideremos el agua, H2O. La
situación es análoga a la del amoniaco, excepto que el oxígeno sólo tiene dos
electrones no apareados, por lo que solamente se enlaza con dos átomos de
hidrógeno, que ocupan dos vértices de un tetraedro; los otros dos están ocupados por pares de electrones no
compartidos (Fig. 1.14).
Según las mediciones, el ángulo H - O - H es de 105º,
menor que el ángulo tetraédrico
calculado y menor aún que el ángulo en el amoniaco. Aquí tenemos dos
voluminosos pares de electrones no compartidos que comprimen los ángulos de
enlace. La longitud del enlace oxígeno-hidrógeno es 0.96 A; se necesitan 118
kcal/mol para romper uno de los enlaces del agua.
Si examinaremos la figura 1.15 podremos ver la gran semejanza que existe entre la forma de las moléculas
de metano, amoniaco y agua, que, debido
a la comparación que hemos utilizado, se debe a la semejanza de los enlaces.
Debido a los pares de electrones no compartidos del
oxígeno, el agua es básica, aunque no tan marcadamente como el amoniaco (sec.
1.22).
Parte 13
1.13 Fuerzas intramoleculares
Debemos recordar que el método específico para la
construcción mental de moléculas que estamos aprendiendo a usar es artificial:
es un proceso puramente intelectual que comprende solapamientos imaginarios de
orbitales imaginarios. Hay otras posibilidades, igualmente artificiales, que
emplean modelos mentales o físicos diferentes. Nuestro conjunto de modelos
atómicos mentales sólo contendrá tres <<clases>> de carbono: tetraédrico
(hibridado sp3), trigonal (hibridado sp2) y digonal
(hibridado sp). Descubriremos que con este conjunto se puede lograr un trabajo extraordinario en la
construcción de cientos de miles de moléculas orgánicas.
Sin embargo, cualquiera que sea el modo de establecerla,
vemos que la estructura verdadera de una molécula es el resultado neto de una
combinación de fuerzas repulsivas y atractivas, que están relacionadas con la
carga y el espín electrónicos.
(a) Fuerzas repulsivas. Los
electrones tienden a mantenerse separados al máximo, porque tienen la
misma carga, y también cuando no están
apareados, porque tienen igual espín
(principio de exclusión de Pauli). Núcleos atómicos de igual carga también se
repelen mutuamente.
(b) Fuerzas atractivas: Los
electrones son atraídos por núcleos atómicos lo mismo que los núcleos por los
electrones debido a su carga opuesta, y
por ello tiende a ocupar la región entre dos núcleos; el espín opuesto permite
(aunque, por sí mismo, no lo estimule
realmente) que dos electrones ocupen la
misma región.
En el metano, por ejemplo, los cuatro núcleos de hidrógeno se hallan separados al máximo. La distribución de los ocho electrones enlazantes es tal que cada uno ocupa la región deseable cerca de dos núcleos - el orbital de enlace - y, sin embargo, exceptuando a su pareja, se sitúa lo más lejos posible de los demás electrones. Podemos visualizar cada electrón aceptado -quizá renuentemente, debido a sus cargas similares - un compañero de orbital con espín opuesto, pero manteniéndose alejado al máximo del resto de los electrones, y aun, como se mueve dentro de los confines difusos de su orbital, haciendo lo posible para evitar la cercanía de su inquieto compañero.
Parte 14
1.14 Energía de disociación de enlace. Homólisis y heterólisis
Hemos visto que cuando se combinan átomos para formar una
molécula se libera energía. Para descomponer una molécula en sus átomos, debe
consumirse una cantidad de energía equivalente. La cantidad de energía que se
consume o libera cuando se rompe o forma un enlace se conoce como energía de
disociación de enlace, D, y es característica del enlace específico. La tabla
1.2 contiene los valores medidos para algunas energías de disociación de enlaces. Puede apreciarse que varía mucho, desde enlaces débiles, como I - I (36 kcal/mol), hasta enlaces muy fuertes, como H - F (136 kcal/mol).
Aunque los valores aceptados pueden variar a
medida que mejoran los métodos
experimentales, hay ciertas tendencias claras.
No debemos confundir la energía de disociación de enlace
(D) con otra medida de fuerza de enlace, llamada energía de enlace (E). Si
comenzamos con el metano, por ejemplo, y rompemos sucesivamente cuatro enlaces carbono-hidrógeno, encontraremos
cuatro energías de disociación de enlace diferentes:
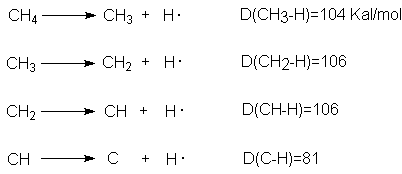
Por otra parte, la energía de enlace carbono-hidrógeno en el metano, E(C - H), es
un solo valor promedio:
![]()
Encontraremos
que, en general, las energías de disociación de enlaces son más útiles
para nuestros propósitos.
Hasta el momento, hemos hablado de romper moléculas en dos átomos, o en un átomo y un
grupo de ellos, de modo que de los dos
electrones que forman el enlace uno se queda con cada
fragmento; esta ruptura de enlace se denomina homólisis. También encontraremos reacciones que implican
ruptura de enlaces de un tipo
diferente, heterólisis, en la que ambos
electrones del enlace quedan en un mismo fragmento.
![]()
(Estas palabras proceden del griego: homo, el mismo; hetero,
diferente, y lisis, pérdida. Para un
químico, lisis significa <<ruptura>> , como, por ejemplo,
hidrólisis, <<ruptura
por agua>>.)
Las energías de disociación de enlaces indicadas en la tabla 1.2 corresponden a homólisis,
por lo que son energías de disociación homolítica de enlaces. Pero
también se han medido para la
heterólisis: algunas de estas energías
de disociación heterolítica de
enlaces se presentan en la tabla 1.3.
Si examinamos estos valores, observaremos que son considerablemente mayores que los de
la tabla 1.2. La heterólisis simple de una
molécula neutra genera, desde luego, un ión positivo y otro negativo. La
separación de estas partículas de carga opuesta consume gran cantidad de
energía: alrededor de 100 kcal/mol más
que la separación de partículas neutras. Por consiguiente, en la fase gaseosa, la disociación de enlaces
generalmente sucede por homólisis, que es la vía más fácil. Pero en un disolvente ionizante (Sec. 6.5) el modo
preferido de ruptura es la heterólisis.
Parte 15
1.15 Polaridad de los enlaces
Aparte de las propiedades ya descritas, algunos enlaces
covalentes tienen otra: la polaridad. Dos átomos unidos por un enlace covalente
comparten electrones, y sus núcleos son mantenidos en la misma nube electrónica. Pero en la mayoría de los casos, estos núcleos no
comparten los electrones por igual: la
nube es más densa en torno a un átomo
que en torno al otro. En consecuencia,
un extremo del enlace es relativamente negativo, y el otro,
relativamente positivo, es decir, se forma un polo negativo y otro positivo. Se dice que éste es un enlace polar o que tienen polaridad.
Podemos indicar la polaridad empleando los símbolos &+
y &-, que indican cargas parciales + y -. (Se dice <<delta más>> y <<delta menos>>.) Por ejemplo:
Cabe esperar que un enlace covalente sea polar si une
átomos que difieren en su tendencia a atraer
electrones, es decir, que
difieren en electronegatividad. Es más, cuanto mayor sea la diferencia en
electronegatividad, más polar será el enlace.
Los elementos más electronegativos son los que se
encuentran en el extremo superior
derecho del sistema periódico. De los elementos que encontraremos en química orgánica, la electronegatividad
más elevada la presenta el flúor, luego el oxígeno, seguido del nitrógeno y el
cloro, a continuación el bromo y, finalmente, el carbono. El hidrógeno no
difiere mucho en electronegatividad del carbono; no se sabe con certeza si es
más electronegativo o menos.
Las polaridades
de los enlaces están íntimamente
ligadas tanto a las propiedades físicas como a las químicas. La polaridad de
los enlaces puede conducir a polaridades
de moléculas, afectando
considerablemente a los puntos de fusión y ebullición, y a la solubilidad. La
polaridad también determina el tipo de reacción que puede suceder en ese
enlace, e incluso afecta a la
reactividad de los enlaces
cercanos.
Parte 16
1.16 Polaridad de las
moléculas
Una molécula es polar cuando el centro de la carga
negativa no coincide con el de la positiva. Tal molécula constituye un dipolo:
dos cargas iguales y opuestas separadas en el espacio. A menudo se usa el
símbolo ® para caracterizar un dipolo, en el que la
flecha apunta desde el extremo positivo hacia el negativo. La molécula tiene un
momento dipolar m, que es igual a la magnitud de la carga, e,
multiplicada por la distancia, d, entre
los centros de las cargas:
m = e x d
en en en
unidades
u.e.s. cm
Debye, D
Es posible medir los momentos dipolares de moléculas por
un método que no puede describirse aquí; algunos de los valores obtenidos se
dan en la tabla 1.4. Nos interesan los valores de los momentos dipolares como
indicaciones de las polaridades relativas de diversas moléculas.
Es un hecho que ciertas moléculas son polares, lo que ha
dado origen a la especificación de que ciertos
enlaces son polares. Nos hemos ocupado primero de la polaridad de los
enlaces simplemente porque es conveniente considerar que la polaridad de una
molécula es una combinación de las polaridades de los enlaces individuales.
Moléculas como H2, O2, N2,
CI2 y Br2 tienen momentos dipolares nulos, o sea, no
son polares. Los dos átomos idénticos
de cada una de estas moléculas tienen, por supuesto, la misma
electronegatividad y comparten
electrones por igual; e es cero y, por consiguiente, también lo es m.
Una molécula como el fluoruro de hidrógeno tiene el
considerable momento dipoalr de 1.75 D.
A pesar de que es una molécula pequeña, el flúor, muy electronegativo,
atrae fuertemente los electrones:
aunque d es pequeña, e es grande y, en consecuencia, m lo es también.
El metano y el tetracloruro de carbono, CCI4,
tiene momentos dipolares nulos. Lógicamente, sería de esperar que los enlaces
individuales al menos los del tetracloruro de carbono fuesen polares, pero
debido a la disposición tetraédrica, altamente simétrica, sus momentos se
anulan (Fig. 1.16). Sin embargo, en el cloruro de metilo, CH3CI, la
polaridad del enlace carbono-cloro no se anula, por lo que tiene un momento
dipolar de 1.86 D. Así, la polaridad de una molécula no sólo depende de la
polaridad de sus enlaces individuales, sino también de sus direcciones, es
decir, de la forma de la molécula.
El amoniaco tiene un momento dipolar de 1.46 D, el cual
podría considerarse como un momento dipolar neto (una suma vectorial) resultante de los momentos de los tres enlaces
individuales, y su dirección sería la indicada en el diagrama. El momento
dipolar de 1.84 D del agua se podría
interpretar de forma similar.
¿Que tipo de momento dipolar cabría esperar para el
trifluoruro de nitrógeno, NF3, que es piramidal como el amoniaco? El flúor es el elemento más
electronegativo de todos por lo que sin duda debería atraer fuertemente los
electrones del nitrógeno; los enlaces N - F deberían ser muy polares y su suma
vectorial debería ser grande, mucho mayor que para el amoniaco, con sus enlaces
N-H moderadamente polares.
¿Cual es la realidad? El trifluoruro de nitrógeno tiene
un momento dipolar de solamente 0.24 D; no es mayor que el del amoniaco, sino
mucho menor.
¿Como podemos explicar esto? Hemos olvidado el par de electrones no compartido. En el NF3
(al igual que en el NH3) este par ocupa un orbital sp3 y
debe contribuir con un momento dipolar en dirección opuesta al del momento neto de los enlaces
N-F (Fig. 1.17);
estos momentos opuestos son casi de la misma magnitud, y
el resultado es un momento pequeño cuya dirección desconocemos. El momento
observado para el amoniaco se debe muy probablemente al par no compartido, aumentado por la suma de los
momentos de enlace. De modo análogo,
los pares de electrones no compartidos del agua deben contribuir a su momento
dipolar y, de hecho, al de cualquier molécula
en al que aparecen.
Los momentos dipolares pueden dar información valiosa
acerca de la estructura de las moléculas. Por ejemplo, pueden descartarse
cualquier estructura para el tretracloruro de carbono que dé lugar a una molécula polar basándose tan sólo
en el momento dipolar, que respalda así la estructura tetraédrica. (Sin
embargo, no la confirma, puesto que se pueden concebir otras estructuras que
también darían como resultado una molécula no polar.)
Los momentos dipolares de la mayoría de los compuestos no
se han medido nunca; para estas sustancias debemos predecir la polaridad a
partir de sus estructuras. Con nuestros conocimientos sobre
electronegatividad podemos estimar la
polaridad de enlace; con los ángulos de enlace
podemos estimar la polaridad de las moléculas, considerando también los
pares de electrones no compartidos.
Parte 17
1.17 Estructura y propiedades físicas
Acabamos de estudiar una propiedad física de los
compuestos: el momento dipolar. También
nos conciernen otras, como los puntos de fusión y ebullición, y la
solubilidad en un disolvente determinado. Las propiedades físicas de un
compuesto nuevo dan indicaciones
valiosas sobre su estructura, y a la inversa, la estructura de una
sustancia a menudo nos dice qué
propiedades físicas esperar de ella.
Al intentar la síntesis de un compuesto nuevo, por
ejemplo, debemos planificar una serie de reacciones para convertir una sustancia que tenemos en la que queremos;
además, debemos desarrollar un método para
separar nuestro producto de
todos los demás compuestos que forman parte de la mezcla reaccionante: reactivos no consumidos,
disolvente, catalizador,
subproductos. Generalmente, el aislamiento y la purificación del
producto consumen más tiempo y esfuerzo
que la propia preparación. La posibilidad
de aislar el producto por
destilación depende de su punto de ebullición
y de los puntos de ebullición de los contaminantes; su aislamiento por
recristalización depende de su solubilidad en varios disolventes y de la de los
contaminantes. El éxito en laboratorio a menudo depende de una adecuada
predicción de propiedades físicas a partir de la estructura. Los compuestos
orgánicos son sustancias reales, no
solamente colecciones de letras escritas sobre un trozo de papel, por lo que debemos aprender a
manejarlas.
Hemos visto que hay dos tipos extremos de enlaces químicos, generados por transferencia de electrones, y covalentes, formado por electrones compartido. Las propiedades físicas de un compuesto dependen en gran medida del tipo de enlaces que mantienen unidos los átomos de una molécula.
Parte 18
1.18 Punto de fusión
En un sólido cristalino las partículas que actúan como
unidades estructurales iones o moléculas se hallan ordenadas de una forma muy
regular y simétrica; hay un modelo geométrico que se repite en el cristal.
Fusión es el cambio desde una disposición muy ordenada de
partículas en el retículo cristalino al más desordenado que caracteriza a los
líquidos (véanse Figs. 1.18 y 1.19). La fusión se produce cuando se
alcanza una temperatura a la cual la energía térmica de las partículas
es suficientemente grande como para vencer las fuerzas intracristalinas que las
mantienen en posición.
Un compuesto
iónico forma cristales en los que las unidades estructurales son iones. El
cloruro de sodio sólido, por ejemplo, está constituido por iones sodio
positivos y iones cloruro negativos que
se alternan de un modo muy regular. Cada ión positivo está rodeado
equidistantemente por seis iones negativos; uno a cada lado, uno arriba y otro
abajo, uno al frente y otro detrás. A su vez, cada ión negativo está rodeado de
forma análoga por seis positivos. No hay nada que podamos llamar molécula de cloruro de sodio; un ión
sodio determinado no <<pertenece>> a ningún ión cloruro en
particular; seis cloruros lo atraen por igual. El cristal es una estructura muy
fuerte y rígida, pues las fuerzas electrostáticas que mantienen a cada ión en
posición son poderosas. Estas poderosas fuerzas interiónicas sólo se superan a una temperatura muy elevada: el
cloruro de sodio tiene un punto de fusión de 801ºC.
Los cristales de otros compuestos iónicos son semejantes
a los del cloruro de sodio, en el sentido de que tienen un retículo iónico,
aunque la disposición geométrica exacta puede ser diferente. En consecuencia,
éstos también tienen puntos de fusión
elevados. Muchas moléculas contienen tanto enlaces iónicos como covalentes: el
nitrato de potasio, KNO3, por ejemplo, está formado por iones K+ y
NO3-; los átomos de oxígeno
y nitrógeno del ión NO3- se mantienen unidos entre sí por
enlaces covalentes. Las propiedades
físicas de compuestos como éste están
determinadas en gran medida por los enlaces iónicos; el nitrato de
potasio tiene aproximadamente el mismo tip de propiedades físicas que el
cloruro de sodio.
Un compuesto no
iónico, aquel cuyos átomos se mantienen unidos entre sí por enlaces
covalentes, forma cristales en los que las unidades estructurales son
moléculas. Para que ocurra la fusión, deben ser superadas que mantienen juntas
a estas moléculas; en general, estas fuerzas intermoleculares son muy débiles,
comparadas con las fuerzas que unen los iones. Para fundir el cloruro de sodio
debemos suministrar energía suficiente para romper los enlaces iónicos entre el
Na+ y el CI-; para fundir el metano, CH4, no
necesitamos suministar energía suficiente para romper los enlaces covalentes
entre el carbono y el hidrógeno, basta con proporcionar energía suficiente para
separar mole´culas de CH4 entre sí. Al contrario que el cloruro de
sodio, el metano se funde a -183ºC.
Parte 19
1.19 Fuerzas intermoleculares
¿Qué tipo de fuerzas mantienen juntas las moléculas
neutras? Al igual que las interiónicas, estas fuerzas parecen ser de naturaleza
electrostática, en las que cargas positivas atraen cargas negativas. Hay dos
clases de fuerzas intermoleculares: interaccione
dipolo-dipolo y fuerzas de Van der Waals.
La interacción
dipolo-dipolo es la atracción que ejerce el extremo positivo de una
molécula polar por el negativo de otras semejante. En el cloruro de hidrógeno,
por ejemplo, el hidrógeno relativamente positivo de una molécula es atraído por
el cloro relativamente negativo de otra:
Como resultado de esta interacción dipolo-dipolo, las
moléculas polares por lo general se unen entre sí más firmemente que las no polares
de peso molecular comparable; esta diferencia entre la intensidad de las
fuerzas intermoleculares se refleja en las propiedades físicas de los
compuestos implicados.
Un tipo de atracción dipolo-dipolo particularmente fuerte
es el enlace por puente de hidrógeno,
en el cual un átomo de hidrógeno sirve
como puente entre dos átomos electronegativos, sujetando a uno con un enlace
covalente, y al otro, con fuerzas puramente electrostáticas. Cuando el hidrógeno
se encuentra unido a un átomo muy electronegativo, la nube electrónica se
distorsiona considerablemente hacia éste, exponiendo el núcleo del hidrógeno.
La fuerte carga positiva del escasamente protegido núcleo del hidrógeno es
atraída por la carga negativa del átomo electronegativo de una segunda
molécula. Esta atracción tiene una fuerza de unas 5 kcal/mol, por lo que es
mucho más débil que el enlace covalente unas 50-100 kcal/mol- que lo mantiene
unido al primer átomo electronegativo. Es, sin embargo, bastante más fuerte que
otras atracciones dipolo-dipolo. En las fórmulas, Los enlaces por puentes de
hidrógeno se indican generalmente por una línea de puntos:
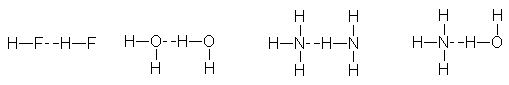
Para
que un enlace por puente de hidrógeno sea importante, ambos átomos electronegativos
deben ser del grupo F, O, N. Sólo es suficientemente positivo un
hidrógeno enlazado a uno de estos elementos y sólo estos tres elementos deben
su efectividad especial a la carga
negativa concentrada sobre sus átomos pequeños.
Deben existir fuerzas entre las moléculas de un compuesto
no polar, puesto que aun estas sustancias se pueden solidificar. Tales
atracciones se conocen como fuerzas de
Van der Waals. Su existencia está
explicada por la mecánica cuántica y podemos describir su origen aproximadamente
como sigue: la distribución promedio de carga en torno a una molécula de metano, por ejemplo, es simétrica, por lo
que no hay un momento dipolar neto. Sin embargo, los electrones se desplazan, de modo que un instante cualquiera
esa distribución probablemente se distorsionará y habrá un pequeño dipolo. Este
dipolo momentáneo afectará a la
distribución de electrones en otra molécula cercana de metano; el extremo
negativo un dipolo de orientación opuesta
en la molécula vecina:
A pesar de que los dipolos momentáneos y los inducidos
cambian constantemente, resulta una atracción neta entre ambas moléculas.
Estas fuerzas de Van der Waals son de muy corto alcance: sólo actúan entre las partes de moléculas diferentes que están en contacto íntimo, es decir,
entre sus superficies. Veremos que la reacción enttre la magnitud de las
fuerzas de Van der Waals y el área de las superficies moleculares (Sec. 3.12)
nos ayudará a comprender el efecto del tamaño y las formas moleculares sobre
las propiedades físicas.
Cada átomo tiene con respecto a otros con los que no esté
unido - ya sea en otra molécula o en otra parte de la misma - un <<tamaño>> efectivo, conocido como
su radio de Van der Waals. A medida
que se acercan dos átomos no alcanzados, aumenta la atracción entre ellos, que
llega al máximo justamente cuando se tocan, es decir, cuando la distancia entre
los núcleos es igual a la suma de los radios de Van der Waals. Si son forzados
a juntarse aún más, la atracción es rápidamente reemplazada por repulsión de Van
der Waals, de modo que los átomos no
alcanzados aceptan juntarse, pero resisten vigorosamente la sobrecarga.
Veremos que las fuerzas de Van der Waals, tanto
atractivas como repulsivas, son importantes para comprender la estructura
molecular.
En el capítulo 6 analizaremos con detalle todas estas
fuerzas intermoleculares, este tipo de enlaces secundarios.
Parte 20
1.20 Punto de ebullición.
Aunque en un
líquido las partículas tienen un arreglo menos regular y gozan de mayor libertad
de movimiento que en un cristal, cada una de ellas es atraída por muchas otras.
La ebullición implica la separación de moléculas individuales, o pares de iones
con carga opuesta, del seno del líquido (Véanse Figs. 1.20 y 1.21). Esto sucede
cuando se alcanza una temperatura suficiente para que la energía térmica delas
partículas supere las fuerzas de cohesión que las mantienen en el líquido.
En el estado líquido, la unidad de un compuesto iónico es de nuevo el ión. Cada ión es retenido
firmemente por varios otros de carga
opuesta. Una vez más, no hay
nada que podamos denominar
realmente molécula. Se necesita mucha
energía para que un par de iones de carga opuesta pueda abandonar el líquido;
la ebullición sólo se produce a temperatura muy alta. El punto de ebullición
del cloruro de sodio, por ejemplo, es de 1413ºC. En el estado gaseoso tenemos
un par iónico, que puede considerarse
como una molécula de cloruro de sodio.
En el estado líquido, la unidad de un compuesto no iónico
es de nuevo la molécula. Aquí, las débiles fuerzas intermoleculares
interaccione dipolo-dipolo y fuerzas de Van der Waals son más fáciles de vencer
que las considerables fuerzas Inter.-iónicas de los compuestos iónicos, por lo
que la ebullición se produce a temperatura mucho más bajas. El metano no polar
hierve a 161.5ºC, y el cloruro de hidrógeno polar a sólo -85ºC.
Los líquidos cuyas moléculas se mantienen unidas por
puentes de hidrógeno se denominan líquidos
asociados. La ruptura de estos puentes requiere una energía considerable,
por lo que un líquido asociado tiene un punto de ebullición anormalmente
elevado para un compuesto de su peso molecular y momento dipolar. El fluoruro
de hidrógeno, por ejemplo, hierve a una temperatura 100 grados más alta que el
cloruro de hidrógeno, más pesado, pero no asociados; el agua hierve a una
temperatura 160 grados más alta que el sulfuro de hidrógeno.
También hay compuestos orgánicos que contienen oxígeno o
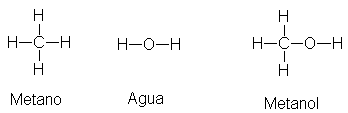
nitrógeno con puentes de hidrógeno Consideremos el metano, por ejemplo, y reemplacemos uno de sus hidrógenos por un grupo hidroxilo, -OH. El compuesto
resultante, CH3OH, es metanol, el miembro más pequeño de la familia
de los alcoholes. Estructuralmente,
no sólo se parece al metano, sino también al agua:
Al igual que el
agua, se trata de un líquido asociado, cuyo punto de ebullición es <<anormalmente>> elevado para un
compuesto de su tamaño y polaridad.
Cuanto más grandes son las moléculas, más fuertes son las
fuerzas de Van der Waals. Se conservan otras propiedades - polaridad, puentes
de hidrógeno -, pero el punto de ebullición aumenta con el tamaño molecular.
Los puntos de ebullición de sustancias orgánicas son bastante más elevados que el de la pequeña molécula no polar del
metano, pero rara vez encontramos
puntos de ebullición por encima de 350ºC; a temperaturas más elevadas, comienzan a romperse los enlaces covalentes dentro de las moléculas, con lo que
compiten la descomposición y la ebullición. Para bajar el punto de ebullición y
así minimizar la descomposición, a
menudo se realiza la destilación de compuestos orgánicos a presión reducida.
Parte 21
1.21 Solubilidad
Cuando se disuelve un sólido o un líquido, las unidades
estructurales iones o moléculas se separan unas de otras y el espacio entre
ellas pasa a ser ocupado por moléculas de disolvente. Durante la disolución,
igual que en la fusión y la ebullición, debe suministrarse energía para vencer
las fuerzas Inter.-iónicas o intermoleculares. ¿De dónde proviene esta energía? La que se requiere para romper los enlaces
entre las partículas del soluto es aportada por la formación de enlaces entre
partículas de soluto y moléculas de disolvente: las fuerzas atractivas
anteriores son reemplazadas por otras nuevas.
Ahora bien, ¿cómo son estos enlaces que se establecen
entre el soluto y el disolvente? Consideremos primero el caso de los solutos iónicos.
Se necesita una cantidad considerable de energía para
vencer las poderosas fuerzas electrostáticas que sostienen un retículo iónico. Sólo
el agua y otros disolventes muy polares
pueden disolver apreciablemente compuestos iónicos. ¿Qué tipo de enlaces
se forman entre iónes y un disolvente polar? Por definición, una molécula polar
tiene un extremo positivo y otro
negativo; por tanto, hay atracción electrostática entre un ión positivo y el
extremo negativo de una molécula de
disolvente, y entre un ión
negativo y la parte positiva de la
molécula de disolvente. Estas atracciones
se llaman enlaces ión-dipolo. Cada uno de estos enlaces ión-dipolo es relativamente débil, pero
en conjunto aportan suficiente energía para vencer las fuerzas
interiónicas del cristal. En la
solución, cada ión está rodeado por muchas moléculas de disolvente, por lo que
se dice que está solvatado; si el
disolvente es agua, se dice que el ión está hidratado.
En solución, tanto en estado sólido como líquido, la unidad de una sustancia
como el cloruro de sodio en el ión, aunque en este caso es un ión solvatado
(véase Figura 1.22).
Para que un disolvente pueda disolver compuestos iónicos,
debe tener también una constante dieléctrica elevada, o sea, debe poseer
propiedades altamente aislantes para disminuir la atracción entre iones de
carga opuesta cuando están solvatados.
El agua debe sus relevantes propiedades como disolvente
de sustancias iónicas, no solamente a su polaridad y asu elevada constante
dieléctrica, sino también a otro factor:
contiene el grupo -OH, por lo que puede formar puentes de hidrógeno. El
agua solvata tanto cationes como aniones; los cationes en su polo negativo
(básicamente, sus electrones no compartidos), y los aniones, por medio de
puentes de hidrógeno.
Pasemos ahora a
la disolución de solutos no iónicos.
Las características de la solubilidad de compuestos no
iónicos están determinadas principalmente por su polaridad. Las sustancias no
polares o débilmente polares se disuelven
en disolventes no polares o ligeramente
polares; los compuestos muy polares
lo hacen en disolventes no polares o ligeramente polares; los compuestos
muy polares lo hacen en disolventes de
alta polaridad. <<Una
sustancia disuelve a otra similar>>, es
una regla empírica muy útil. El metano
es soluble en tetracloruro de carbono, porque las fuerzas que mantienen
unidas las moléculas de metano y las de tetracloruro de carbono - las
interacciones de Van der Waals -
son reemplazadas por otras muy
similares, las que unen moléculas de tetracloruro de carbono a moléculas de metano.
Ni el metano ni
el tetracloruro de carbono son apreciablemente solubles en agua, cuyas moléculas, muy polares, se atraen mutuamente
por interacciones dipolo-dipolo muy intensas: los puentes de hidrógeno; por otra parte, sólo podría haber fuerzas atractivas muy débiles entre las moléculas de agua y las no polares
de metano o de tetracloruro de carbono.
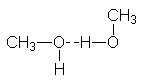
Por el contrario, el metanol, CH3OH, compuesto orgánico muy polar, es totalmente
soluble en agua. Los puentes de hidrógeno entre las moléculas de agua y las de
metanol pueden reemplazar fácilmente a los puentes de hidrógeno similares formados
entre diferentes moléculas de metanol y diferentes moléculas de agua.
La comprensión de la naturaleza de las soluciones es
fundamental para entender la química orgánica. La mayoría de las reacciones
orgánicas se efectúan en solución, y es cada vez más evidente que el disolvente
hace mucho más que simplemente unir moléculas diferentes para que puedan
reaccionar entre sí. El disolvente está implicado en las reacciones que tienen lugar en él; cuánto y en qué forma
está implicado es algo que empieza a saberse ahora. En el capítulo 6, cuando
estudiamos un poco más las reacciones orgánicas y cómo se realizan, volveremos
sobre este tema - que apenas tocamos aquí y examinaremos en detalle la función del disolvente.
Parte 22
1.22 Acidos y bases
Al pasar ahora de las propiedades físicas a la químicas,
revisemos brevemente un tema conocido que es fundamental para la comprensión de
la química orgánica: acidez y
basicidad.
Los términos ácido
y base se han definido de varias formas, correspondiendo cada
definición a un modo particular de
considerar las propiedades de acidez y basicidad. Nos será útil observar ácidos
y bases desde dos de estos puntos de vista; el que elijamos dependerá
del problema que se tenga a mano.
De acuerdo con la definición de Lowry-Bronsted, un ácido es
una sustancia que entrega un protón, y una base, una que lo acepta. Al disolver ácido sulfúrico en agua, el
ácido H2SO4 entrega un protón (núcleo de hidrógeno) a la
base H2O para formar el nuevo ácido H3O+ y la
nueva base HSO4-. Cuando el cloruro de hidrógeno
reacciona con el amoniaco, el ácido HCI
entrega un protón a la base NH3 para formar el nuevo ácido NH4+
y a la nueva base CI-.
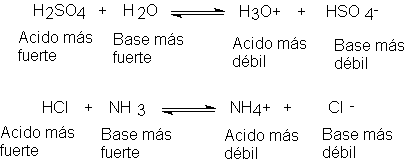
Según la definición de Lowry-Bronsted, la fuerza de un ácido
depende de su tendencia a entregar un protón, a la de una base, de su tendencia
a aceptarlo. El ácido sulfúrico y el cloruro de hidrógeno son ácidos fuertes,
puesto que tiende a entregar un rpotón con mucha facilidad; a la inversa, el ión bisulfato, HSO4-,
y el ión cloruro deben ser necesariamente bases débiles, puesto que demuestran
poca tendencia a adherirse a protones. En las dos reacciones que acabamos de
describir, el equilibrio favorece la formación del ácido y la base más débiles.
Si se mezclan H2SO4 y NaOH acuosos,
el ácido H3O+ (ión hidronio) entrega un protón a la base
OH- para formar el nuevo ácido H2O y la nueva base H2O.
Al mezclar NH4CI y NaOH acuosos, el ácido NH4+
(ión amonio) entrega un protón a la base OH- para formar
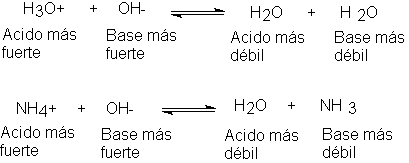
el nuevo ácido H2O y la nueva base NH3.
En ambos casos, la base fuerte, el ión hidróxido, ha aceptado un protón para formar el ácido débil H2O.
Si disponemos estos ácidos en el orden indicado, debemos disponer
necesariamente las bases (conjugadas) correspondientes en orden opuesto.
Fuerza
ácida H2SO4 > H3O+ > NH4+ > H2O
HCI
Fuerza
básica HSO4- < H2O < NH3
< OH-
CI-
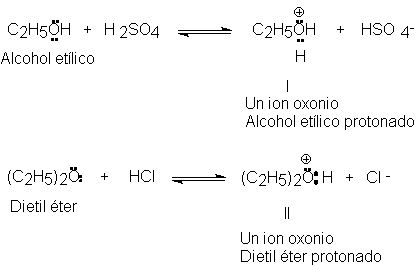
Al igual que el agua, muchos compuestos orgánicos que
contienen oxígeno pueden actuar como bases y aceptar protones; el alcohol
etílico y el dietil éter, por ejemplo, forman los iones oxonio I y II. Por conveniencia, a menudo nos referimos a una
estructura del tipo I como un alcohol protonado, y a una del tipo II,
como un éter protonado.
Según la definición de Lewis, una base es una
sustancia que puede suministrar un par de electrones para formar un enlace
covalente, y un ácido, una que puede recibir un par de electrones para formar un enlace covalente. De este
modo, un ácido es un aceptor de pares
de electrones, y una base, un donante
de pares de electrones. Este es el más fundamental de los conceptos
ácido-base, y también el más general, ya que incluye todos los demás conceptos.
Un protón es un ácido, pues es deficiente en electrones y necesita un par de
ellos para completar su capa de valencia. El ión hidróxido, el amoniaco y el
agua son bases, pues tienen pares de electrones disponibles que pueden compartir. En el trifluoruro de boro, BF3,
el boro sólo tiene seis electrones en su capa externa, por lo que tiende a aceptar otro par para completar su octeno.
El trifluoruro de boro es un ácido, y se combina como bases como el amoniaco o
el dietil éter.
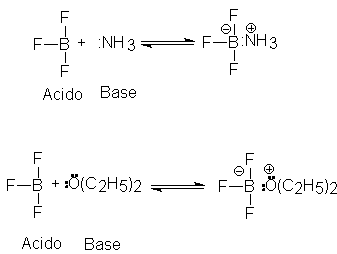
El cloruro de aluminio, AICI3, es un ácido por
la misma razón. El cloruro estánnico, SnCI4, tiene un octeto
completo en el estaño, pero puede aceptar pares de electrones adicionales (por
ejemplo, en SnCI62-), por lo que también es un ácido.
Indicamos una
carga formal negativa sobre el boro en estas fórmulas porque tiene un electrón
más uno del par compartido con oxígeno o nitrógeno de lo que puede neutralizar por medio de su carga nuclear;
correspondientemente, se indica el nitrógeno u oxígeno con una carga formal positiva.
Encontraremos que el concepto de Lewis de ácidos y bases
es fundamental para la comprensión de la química orgánica. Para dejar bien claro que hablemos de este tipo
de ácido o base, emplearemos a menudo
la expresión ácido de Lewis (o base de Lewis) o, a veces, ácido (o base) en el sentido de Lewis.
Al igual que las físicas, las propiedades químicas
dependen de la estructura molecular. ¿Cuáles son las características de la estructura de una molécula que nos
permite diagnosticar su carácter ácido
o básico? Podemos intentar contestar a esta pregunta ahora de una forma
general, aunque más adelante volveremos a ella muchas veces.
Para ser ácida en el sentido de Lowry-Bronsted, una
molécula debe contener, desde luego, hidrógeno. En gran medida, el grado de acidez
lo determina la clase de átomo unido al
hidrógeno y, en particular, la
capacidad de ese átomo para acomodar el par de electornes que el ión hidrógeno
saliente deja atrás. Esta capacidad
parece depender de varios
factores, los que incluyen (a) la electronegatividad del átomo, y (b) su tamaño. Así, dentro de un periodo
determinado de la tabla periódica, la acidez aumenta con el aumento de la
electronegatividad:
Acidez
H-CH3 < H-NH2 < H-OH < H-F
H-SH < H-CI
Y dentro de un grupo determinado, la acidez aumenta con
el tamaño:
Acidez
H-F < H-CI < H-Br < H-I
H-OH < H-SH< H-SeH
Entre los compuestos orgánicos, puede esperarse que
tengan una acidez de Lowry-Bronsted apreciable aquellos que contienen los
grupos O - H, N - H y S - H.
Para que una molécula sea ácida en el sentido de Lewis, debe ser deficiente en
electrones; en particular, buscaríamos en ella un átomo con sólo un sexteto
electrónico.
Para ser básica, tanto en el sentido de Lowry-Bronsted
como en el de Lewis, una molécula debe disponer de un par de electrones para
compartir. Su disponibilidad está determinada en gran medida por el átomo que
los contiene: su electronegatividad, su tamaño y su carga. La función de estos factores es aquí necesariamente
opuesta a lo que hemos observado para la acidez: cuanto mejor acomode el átomo al apr de electrones, menos
disponibles estará éste par ser compartido.
Parte 23
1.23 Isomería
Antes de comenzar el estudio sistemático de las distintas
clases de compuestos orgánicos, veamos un concepto adicional que ilustra
particularmente bien la importancia fundamental de la estructura molecular: el
concepto de isomería.
El compuesto alcohol
etílico es un líquido que hierve a 78ºC. Su análisis (por métodos que se
describen más adelante, sec. 2.27)
demuestra que contiene carbono,
hidrógeno y oxígeno en la proporción 2C:6H:IO. Su espectro de masas indica que su peso molecular es 46, por lo
que su fórmula molecular debe ser C2H6O. Es un conpuesto
bastante reactivo; por ejemplo, si se
deja caer un trozo de sodio metálico en
un tubo de ensayo que contiene alcohol etílico, se produce un burbujeo vigoroso y se consume el sodio; se desprende
hidrógeno gaseoso y queda un compuesto de fórmula C2H5ONa. También reacciona con ácido
yodhídrico para formar agua y un compuesto de fórmula C2H5I.
El compuesto dimetil
éter es un gas con punto de ebullición de -24ºC. Es, evidentemente, una
sustancia diferente del alcohol
etílico: no sólo difiere en sus propiedades físicas, sino también en las químicas. No reacciona
con el sodio metálico. Como el alcohol etílico, reacciona con el ácido yodhídrico, pero da un compuesto de fórmula CH3I. El análisis del
dimetil éter indica que contiene carbono, hidrógeno y oxígeno en la misma
proporción que el alcohol etílico, 2C:6H:IO; tiene el mismo peso molecular, 46.
Concluimos que tiene la misma fórmula molecular, C2H6O.
Tenemos aquí dos sustancias, alcohol etílico y dimetil
éter, que tienen la misma fórmula molecular, C2H6O, y,
sin embargo, son dos compuestos claramente diferentes. ¿Cómo podemos explicar
su existencia? La respuesta es que
difieren en su estructura molecular. El alcohol etílico tiene la estructura representada por I y el dimetil éter
la representada por II. Veremos que las diferencias en propiedades físicas y
químicas de estos dos compuestos pueden explicarse fácilmente a partir de sus
diferencias estruturales.
Los compuestos
diferentes que tienen la misma fórmula molecular se llaman isómeros ( del
griego: isos, igual, y meros, parte). Contienen igual número de las mismas clases
de átomos, pero éstos están unidos entre sí de manera distinta. Los isómeros
son compuestos diferentes, porque
tienen estructuras moleculares distintas.
Esta diferencia en estructura molecular genera
propiedades distintas; son estas diferencias las que nos revelan que estamos
tratando compuestos diferentes. En algunos casos, la diferencia estructural y
por consiguiente las propiedades
distintas es tan marcada que los isómeros se clasifican en familias
químicas diferentes como, por ejemplo, alcohol
etílico y dimetil éter. En otros
casos, la diferencia estructural es tan sutil que sólo puede describirse en
función de modelos tridimensionales. Entre estos dos extremos hay otros
tipos de isomería.